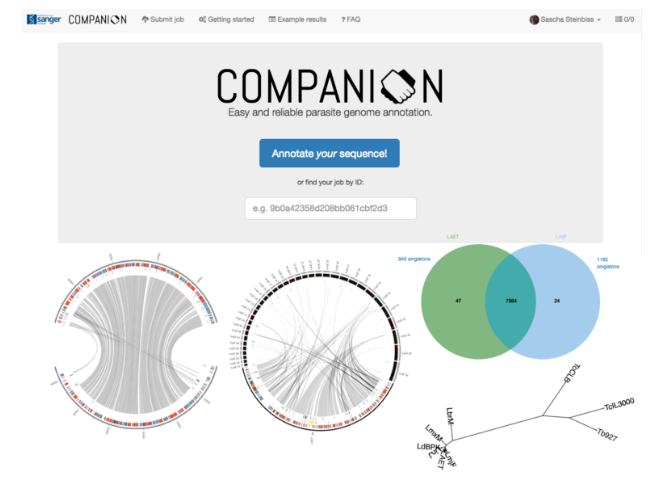
Companion:一种新的寄生虫基因组生成和可视化注释工具
如果大量的寄生虫基因组是为了对抗疾病和生产疫苗和药物而产生的,但是没有人能比较这些基因组会发生什么?Sascha Steinbiss&Thomas Otto在这个博客中回答了这个问题,最初发布在Wellcome Trust Sanger Institute网站上。
在过去的十年里,基因组测序和组装技术有了巨大的进步,到目前为止,研究人员能够在较短的时间内以相对较低的成本获得细菌和小型真核生物近乎完美的基因组序列。即使对小型研究实验室来说,廉价制作好的组装也能使测序民主化,并产生许多新的基因组草案,包括各种新的寄生虫基因组。这一趋势反映在序列装配可用工具的数量不断增加(到2016年超过60个)
然而,为这些生物体生成高质量的标准化注释,即基因的位置和功能以及其他相关特征的问题仍然存在。详细和完整注释的可用性是使随后的比较跨物种分析能够识别单个物种或菌株之间差异的关键。
这种差异的例子可能是共同和/或物种特定基因和功能的丧失或获得。在细菌世界中,用于快速注释基因组的软件工具存在,但到目前为止,寄生虫的等效性缺失。
介绍一种新的软件工具
为了满足这一需求,我们开发了Companion,一个新的软件工具和web服务器,利用我们已经知道的有关物种的信息,在很短的时间内生成寄生虫基因组的全面注释。
Companion的独特功能包括可视化组装质量、将基因内容与参考基因组进行比较,以及提供可轻松提交到欧洲核苷酸档案(ENA)等公共数据库的文件。
赋予数据科学意义
但是,即使一个草案汇编是可用的,没有注释,它仍然是一个不可理解的数据串,没有科学意义。要利用它,我们需要知道蛋白质编码基因和非编码基因的位置,以及它们的功能是什么。
尽管发现这些基因是一个古老的挑战,有专门的工具可供使用,但基因发现的任务仍然是一个开放的问题。不完美的注释,即缺失、错误或仅部分描述的基因模型以及错误的功能关联,可能会严重影响任何类型的下游分析。
然而,现实已经表明,最好的结果只有通过并行使用多个工具,然后再进行手动校正才能获得。
最后,虽然需要发布,但向ENA这样的数据库提交注释文件通常是一个挑战,因为必须遵循特定的命名法。
如何使用?
为了帮助寄生虫社区克服这些问题,我们开发了同伴(综合寄生虫注释)软件,作为一个免费的公共资源。虽然它主要作为一个web服务器提供,但也可以在本地安装它来注释不能在线运行的基因组。
对于寄生虫学家的主要目标受众,我们提供了以前无法比拟的简单注释:只需上传程序集,从我们的62个寄生虫基因组中选择一个相关的参考物种,然后按下按钮。
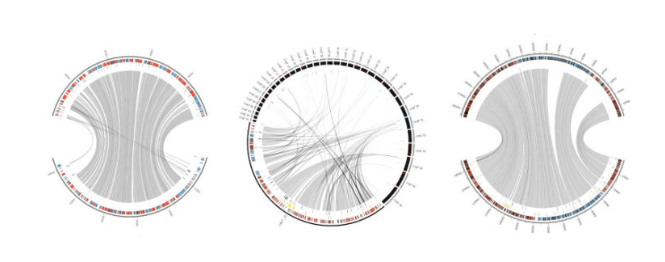
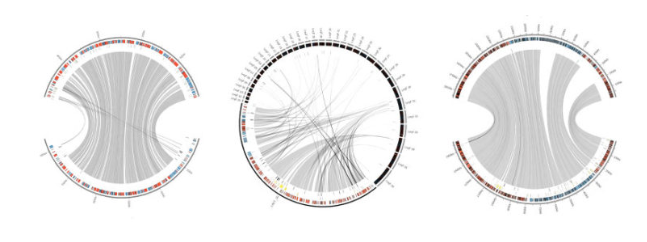
4-6小时后(取决于组装质量和参考尺寸),将发送一封电子邮件,指示用户进入其注释基因组。Companion提供了基本的统计数据,如基因数量、基因密度、每个T、C、G的比例以及DNA中的一个碱基等,但更有趣的是,还提供了第一个比较结果,如说明新注释物种与其他物种关系的系统发生树,或与参考物种相关的基因含量。此外,在自动生成的圆形图中,可以很容易地观察到装配的质量以及大规模的重新排列。如果用户对结果满意,现在可以很容易地将其上传到ENA,这个过程过去是另一个很大的努力。当然,Companion生成的注释也可以作为后续手动策展的良好起点。
第一个主要的用例是使用Companion注释各种新的动质体基因组,包括12个利什曼原虫和锥虫,以及Crithidia和endorypanum基因组,其中大多数可从TriTrypDB获得。
自2016年初Companion公开发布以来,我们已经统计了来自世界各地的120多条注释运行,并且越来越受欢迎。此时,我们要感谢Wellcome Trust Sanger Institute infrastructure systems团队维护服务器。Companion已经被证明是多功能的:尽管Companion的主要目的是注释整个基因组,但用户报告说,他们有时只将其用于伪染色体连合组件,这一功能作为web应用程序也是很少见的。
Companion是使用最先进的技术实现的:Nextflow工作流管理系统来协调管道,GenomeTools基因组分析工具包用于低级脚本,Rails开发生态系统用于web服务器。所有代码都可以在免费的开源许可下使用。
总之,companion正在生成一个高质量的草稿注释,可以很容易地提交给数据库,使社区能够从这些已测序的寄生虫中学习。它还提供了各种输出,允许用户最终将新注释的基因组与参考基因组进行比较,可能为进一步的研究指明了第一个方向。