短串联重复序列 PCR,由于 STR-PCR 基因座仅需少量模板 DNA, 灵敏度比传统的 DNA 指纹高很多,而且 STR-PCR 扩增片段长度较 短,一般为 100〜400bp,对于法医常遇到的仅含降解 DNA 的陈旧性斑痕,STR-PCR 比传统的 DNA 指纹灵敏度和准确度更高。因此,STR 分型被认为是第二代法医 DNA 指纹技术的核心, 是目前国内外法医学个体识别和亲权鉴定的主要技术发展方向。
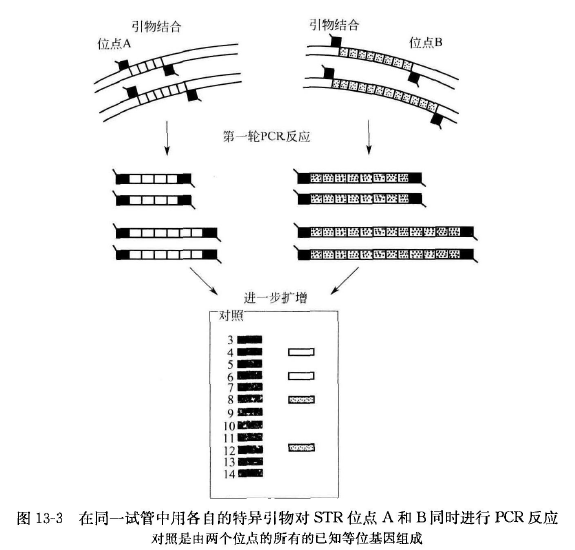
短串联重复序列 PCR 的基本原理是在人类基因组中,STR 是高度多态性标记的丰富来源,可采用 PCR 反应检测。目前 STR-PCR 技术已形成多位点检测方法,即在同一分析反应试管中同时扩增来自两个或更多的位点的等位基因(图 13-3), 扩增的重复序列由于重复次数的差异导致 STR 基因座的等位基因分型不同,在电泳分离后,用放射性同位素、银染或荧光检测可区别不同的基因型。检测时需注意:
① PCR 的模板 DNA 区域必须包括 STR DNA 的侧翼区序列,DNA 总长度为 100〜400bp;
② 进行多基因座同时扩增时,根据不同的基因座序列设计不同的特异引物,特异引物可采用荧光素标记(FL)、梭基-四甲基罗丹明标记(TMR)或 6-羧基-4',5'-二氯-荧光素(JOE)等标记;
③ 在单管中对所有基因座同时扩增后,通过单一进样或单一胶道在遗传分析仪及 DNA 测序 仪上进行电泳分析。
与扩增片段长度多态性 (amplified fragment length polymorphism, AMP-FLP) 和可变数目串联重复序列 (variable number of tendem repeat, VNTR) 等分型方法相比,STR 基因分型方法所扩增的产物长度小得多 (小于 500bp), 因此更适合于对降解的 DNA 模板进行分析。此外,STR 分型适用于多种 DNA 纯化方法得到的 DNA, 这些 DNA 的量往往较少而不够作 Southern 印迹分析。
STR-PCR 产物具有不连续的可分离的长度,可以用每个基因座的几个或所有等位基因的片段构建成等位基因阶梯 (allelic ladder), 肉眼观察或利用仪器比对同一基因座的等位基因阶梯和扩增样品,可快速和准确地确定等位基因座。试验中仔细选择的 STR 基因座和引物可以避免或减少伪带的产生,包括与 RqDNA 聚合酶相关的影子带及末端核昔酸的减少。影子带,有时称为「n-4 带型」、「鬼带」或「暗带」,是由于 DNA 扩增中缺失了一次重复,或样品中 DNA 本身的改变,或二者的结合,它的出现主要与基因座本身和重复的 DNA 序列有关;通常的 PCR 反应中,由于 Taq DNA 聚合酶以不依赖于模板的方式会在扩增的 DNA 片段的 3』末端添加核苷酸,一般是腺苷酸,发生这一现象的频率取决于不同的引物序列;STR-PCR 产物有时常观察到比预计少一个碱基的假带 (无末端添加),因此将引物序列进行修饰,并在扩增流程的最后 一步,加入 60°C 延伸 30 min 的步骤,可使在所用的 DNA 模板上得到完全的末端核苷酸添加。 此外,微变化等基因座 (基因座间相差的长度不是重复次数所致) 的存在常使等位基因座的解释 和确认变得复杂,这和高度多态性、趋向微变化以及突变率的增加相关。
短串联重复序列 PCR 的基本过程可分为如下几步:
1. DNA 的提取
使用螯合树脂 (chelex resin) 可一步完成 DNA 的提取和变性,碱性螯合树脂和高温可以裂 解细胞并使 DNA 变性。对于大多数体液斑点来说 (尤其是对于那些遭受到不利条件的),在提 取 DNA 之前应进行水洗。首先将样品在 5% 螯合树脂悬浮液中煮沸,离心后取上清提取 DNA 或 直接 PCR。DNA IQ™ System (DC6700) 是专门为法医及父权鉴定中 DNA 样本分离及定量而设计的新技术,主要是使用磁性颗粒简便有效地制备来源于斑迹、血液、溶液等用于 STR 分析的样本。DNA IQ™ System 树脂提取 DNA 可消除案件中经常遇到的 PCR 抑制物及污染物。
2.DNA 的定量
扩增出适宜的 DNA 量对获得令人满意的结果是非常重要的。如果起始材料太少只产生部分结果;起始材料太多则产生非特异的扩增产物。在有如细菌 DNA 污染存在的情况下,必须能测 到 ng 级浓度的人类 DNA。狭缝印迹方法被广泛用来确定特异性灵长类 DNA, 它是一种夹心方 法,将提取的样品固定在尼龙条上,然后加入一个 40bp 的对灵长类 DNA 序列特异性的生物素 标记的探针,再加入结合链霉亲和素的过氧化物酶进行酶催化的颜色反应,反应中以过氧化物酶 作为反应的底物。颜色反应的强度与一系列的对照进行比较可以估计提取物中 DNA 的浓度,Pi�coGreen 定量法也适合于对 STR 分析中小量样品的 DNA 浓度进行定量。通常 DNA 样品的最终 浓度达到 1〜10ng/μl 即可检测。
3. DNA 的扩增
(1) PCR 扩增体系
10XPCR 缓冲液 2. 5μl 模板 DNA(5〜10ng/μl) 2. 5μl
2. 5 mmol/L 的 dNTP 混合物 1μl ddH2O 15. 7μl
引物混合物 (l0μmol/L) 2. 5μl 总反应体积 25μl
Tag DNA 聚合酶 (5U/μl)0. 8μl
(2) PCR 扩增程序 用 PE GeneAmp PCR System 9700 热循环仪的扩增程序:
95°C llmin, 96°C 2 min, 94°C lmin, 60°C lmin, 70°C 1. 5 min, 10 个循环; 90°C lmin,60°C 1 min, 70°C 1. 5 min, 18〜22 个循环;60°C 30 min。4°C 保存。
使用热循环仪可迅速准确改变温度。首先样品在高温下变性,产生的单链 DNA 可以作为模 板,然后降低温度,使每条引物与靶序列的侧翼区域退火。再将温度调到&QDNA 聚合酶催化 延伸反应所需的最佳温度维持 60s。
温度循环的次数取决于被扩增的位点,通常是随试验而调整的。同样,退火温度受引物设计 以及选择单位点扩增还是复合扩增的影响,当用例如 ABI377 荧光检测系统进行多位点 STR 分析 时,引物采用不同的染色标记以便把每个位点的等位基因区分开来,使 DNA 片段大小不重叠, 从而提供准确的结果。
4. 产物分离
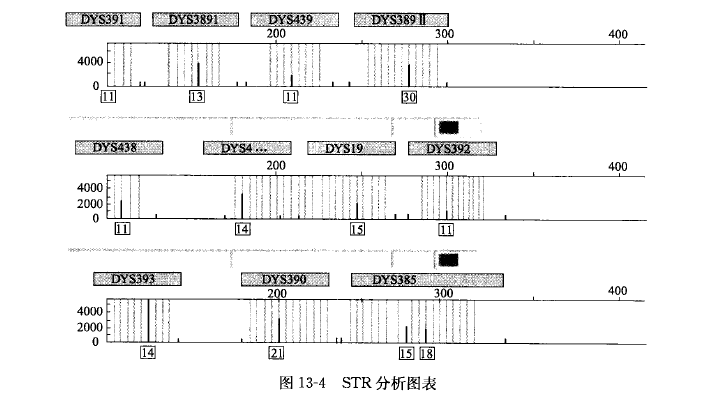
最普遍的分离 PCR 产物的方法是在 ABI PRISM 377 上使用自动荧光检测的电泳。这种基因 测序仪能对经电泳分开后的 PCR 产物进行激光扫描。样本经过 PCR 扩增后,采用 ABI PRISM 377 DNA 测序仪进行检测,通过将参照物和 DNA 分子量标准(具有不同的颜色)进行同步电 泳,并由计算机分析软件分析,就可以对 DNA 片段进行很准确的测定(图 13-4), 准确性通常 能达到区分相差一个碱基对的片段,将扩增样本片段、等位基因阶梯和内标进行比较,从而达到 分型的目的。
内标(ILS)包含多个 DNA 片段,如 Promega 公司的 PowerPlex 16 系统中内标 600(ILS600)含 22 个 DNA 片段,长度分别为 60bp、80bp、lOObp. 120bp、140bp、160bp. 180bp. 200bp、 225bp、 250bp、 275bp、 300bp、 325bp、 350bp、 375bp、 400bp、 425bp、 450bp、 475bp、 500bp、550bp 及 600 bp。这些片段用 CXR 标记,可以在 ABI PRISM310. ABI PRISM3100 测序 仪及 ABI PRISM377 测序仪上同 STR-PCR 的扩增产物一起检测。ILS 用在每一个样本中可以提 高 STR-PCR 扩增产物的精确度。
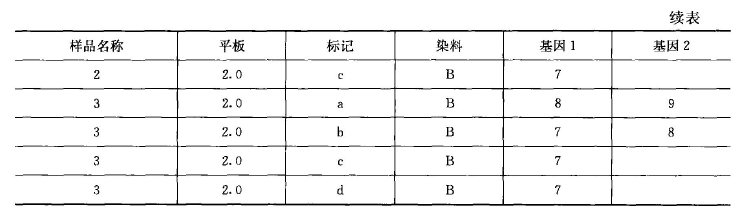
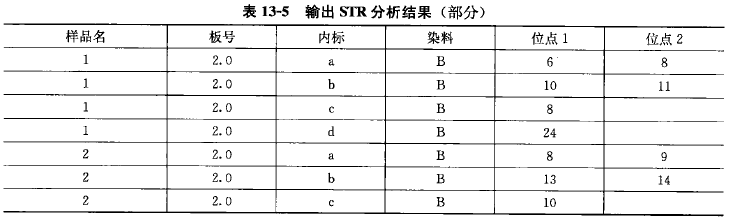
STR 基因分型:带有荧光标签的 PCR 引物在 STR 位点进行 PCR 扩增后,在 ABI3100 测序 仪上检测会产生相应等位基因的峰,通过 GENEMAPPER SOFTWARE 分析,这些峰会显示等 位基因的大小和强度(图 13-4), 并以表格的形式输出分析结果(表 13-5)o 通常 STR 的分型在 多样品检测中能得到准确的结果,当检测 1000 个来自全血的 DNA 样品时,968 个样品能产生清 楚的峰和确切的 STR 位点鸣叫,32 个样品由于低质量的 DNA 失败(ACGTInc)。
5. 数据库
许多年来法医学家一直有一个目标,就是能建立一个包含欧洲各国家的法医学数据库,STR 技术的引进有助于促进这一数据库的建立,目前已经成立了一些组织,它们的任务是为 STR 位 点的使用提出建议。STR 技术的使用需要考虑许多标准,其中包括确保精确结果能够相互交换 的各种合法的系统和标准。个人 DNA 图谱被收集并储存在计算机数据库中的程度取决于当地的 立法情况,并且决定了所建立的数据库的效力。
在英国,尽管依据单位点探针(single locus probe, SLP)结果的数据库已经形成,但是在 1995 年颁布了一个综合性的法律之后,才形成了真正的第一个全国性 DNA 数据库。使用 STR 复合扩增建立个人图谱是形成这个 DNA 索引系统的基础(示意图见图 13-5)。当决定选用哪一个组分的 STR 时必须考虑以下准则:
①每一个位点的等位基因数目;
②复合扩增成功的能力(无偏性扩增);
③尽可能少的人工产物(断断续续和错误的峰);
④每一个位点应该位于不同的染色体上;
⑤应该能得到每个位点的等位基因在群体中的频 率分布资料;
⑥结果易于解释;
⑦试剂必须易于获得。
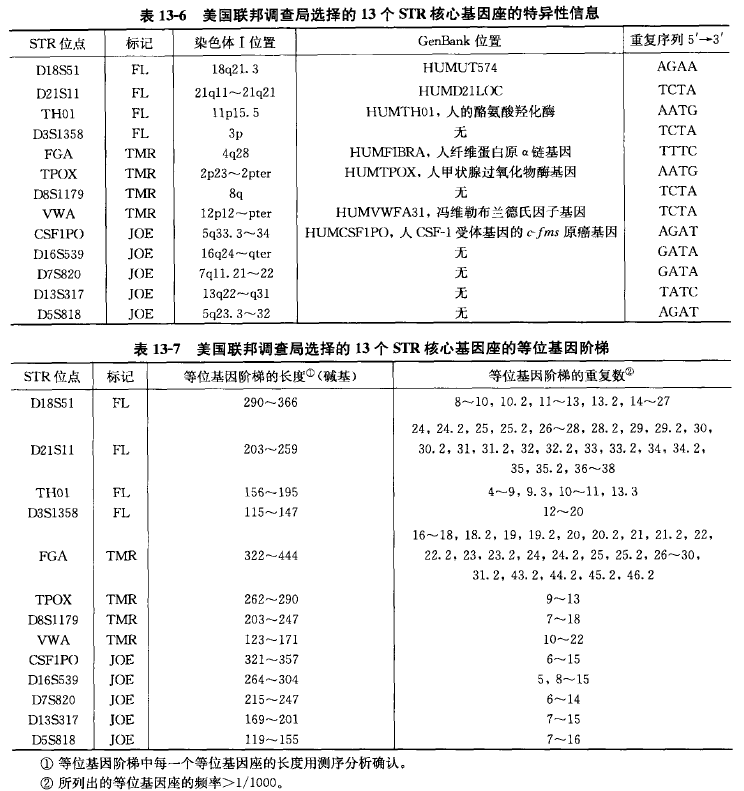
近年来欧洲实验室的研究表明,除在 Y 染色体上 的位点外经常使用的 STR 位点有 50 多个。起初,英国 的数据库是围绕 4 个 STR 位点的复合扩增的结果设计 的,即 THO1、VWA、FES 和 F13A。以后发展了复合 扩增的 6 个 STR 位点——THO1、VWA、FGA、 D8S1179. D18S51 和 D21S11。1999 年复合扩增的位点 增加到 10 个,称为第二代复合扩增位点(SGM),被英国国家 DNA 数据库采纳,应用于日常案件,并且 在欧洲的许多实验室得到广泛使用。在由国际刑警组 织注册的对性侵犯定罪的立法提案程序中,所有参与 的实验室都必须检测 4 个重要位点,即 THO1、VWA、FGA 和 D21S11, 它们的个体识别能力强而有效,已成为欧洲众所周知的核心位点,1999 年后又扩展了另外的 3 个位点。最近在北美已经引入以 STR 为基础的 DNA 索引数据库,建立了 13 个 STR 位点的复合扩增:THO1、VWA、 FGA、TPOX、CSF1PO. D3S1358、D5S181、D3S820、D8S1179. D13S317. D16S539、D18S51 和 D21S11。由于法医科学实验室对 STR 复合扩增具有极大的兴趣以及它的商业潜能,目前已经 有市售的包含 SGM 和一些北美复合扩增位点的试剂盒。美国联邦调査局(FBI)选择 13 个 STR 核心基因座的分型数据输入到 CODIS(联合 DNA 索引系统,即美国犯罪人员分型数据库)中(表 13-6 和表 13-7)。
6. 结果解释
在给所有法医鉴定证据的同时,必须给出一些匹配显著性的信息,这也是 SLP 技术中对 DNA 图谱分析有争议的方面。由于带的大小范围较大(大约 2〜20kb)以及不同样品的电泳迁 移率具有轻微的漂移,所以要确定匹配,必须在实验的基础上确定一个容许「视窗」。一旦一个 匹配被确定,必须同时给出总带模式的许多值。各种等位基因出现的频率参照群体频率数据库进 行计算。为了使每个等位基因的计算频率增加,获得对这个图谱稀有特征的整体估计,使各位点 在染色体间和染色体内保持独立性非常重要,美国国立科学院目前正在研究关于推荐给法庭处理 数据的最佳方法。过去,在获得匹配图谱以后,就直接给出一个可能性的证据。现代统计学 更偏重于使用以 Beyesian 统计方法为基础的或然率(likelihood ratio). 在这种方法中后概率(犯罪概率)是对所有的证据进行评价后而得到的前概率中得来的。DNA 的结果将形成前概率(pri�or odds)的一部分。然而,在最近上诉的一个法庭审判中则认为:对于给定的特异的基因型观察,法医科学家应该坚持鉴别统计学。
计算机分析软件在 STRs 技术中的应用,使分辨率达到可以区分一个碱基对的差异,通过选 择位于不同染色体上的 STRs 位点以保证遗传的独立性,从而精确估计等位基因并进行分型,在 法医学中具有很强的鉴别能力。