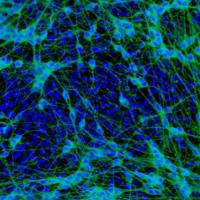
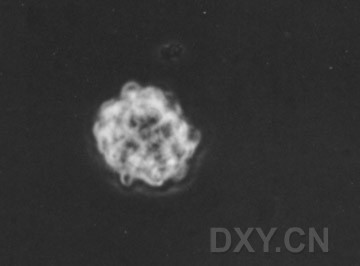
原理

材料与仪器
步骤
材料




步骤
一、胚胎和成体CNS部位的解剖
有关胚胎和成年大鼠的海马和脊髓组织的解剖在这里只作简单介绍,更为详细的实验介绍可参见第十四章。
胚胎CNS
1.腹腔注射麻醉剂使孕鼠深度麻醉,复合麻醉剂成分为:盐酸氯胺酮(ketamine,44 mg/kg),马来酸乙醜丙嗪(acepr〇mazine,4?Omg/kg)和甲苯噻嚷(romPun,0.75 mg/kg)。
2.切开腹部,取出子宫双角,放置冰上,从羊膜囊内取出胚胎置于冷的D>PBS中。
3.测量胚胎的顶-臀长度,以确定胎龄。
海马
1.取下胚胎大脑并置于D-PBS中。用镊子固定胎脑,从中线向一侧撕开皮层并将其平放,用尖头镊子夹住组织,在皮层下方,用弯头眼科剪切下海马。
2.于显微镜下,将海马组织转入D-PBS中。
3.从对侧取下海马,用尖头镊子剥去附着的脑膜和血管组织,收集并混合15~20个胚胎的海马。
脊髓
1.于显微镜下,使胚胎侧卧于无菌的I>PBS中,用眼科剪从椎管侧面剪开,并在对侧做一同样切口,在颈段和腰部下端离断脊髓。
2.取出脊髓,并剥离周围附着的组织,收集并混合15~20个胚胎的脊髓。
成体中抠神经系统海马
1.将成年(3~6个月)雌性Fisher344大鼠麻醉,断头处死,取出大脑并置于含有冷的D-PBS的平皿中。
2.在显微镜下切断胼胝体,于丘脑上方分离左、右大脑皮质,向后翻起大脑皮质半球的后缘,沿两侧海马的内外侧缘分离穹隆和海马辙,取出海马。
成体脊髓
将大鼠麻醉,使其侧卧,从侧面切开椎管,从对侧做一同样切口,小心取出脊髓,剥离附着于脊髓上的结缔组织,于显微镜下,分别解剖出骶、胸、腰、颈段。
二、胚胎中枢神经系统区域的原代培养的建立

三、成体CNS 来源的神经干细胞培养系统的建立


四、克隆培养细胞的分离

五、干细胞的免疫细胞化学染色的鉴定(Petersonetal.1996)


六、干细胞的分化

七、用立体定位移植法将干细胞植入成年鼠脑
待移植动物的准备步骤与第十四章所介绍的胚胎细胞移植相似。此外,有关移植的步骤、动物的灌注、脑组织的切片以及用组织化学和免疫细胞化学染色方法对移植细胞的鉴定等一些更为详细的实验步骤可参见Suhonen等(1997)的实验。以下将这些方法作简单介绍。
移植细胞的制备

神经千细胞在成年大鼠脑中的移植

八、成年大鼠的灌注


九、大鼠脑切片
1.为便于在冰冻切片机上切片,应尽可能将包含所需靶区域的脑组织块修剪至最小。
2.将修剪好的脑组织用OCT包埋并固定在冰冻切片机的夹具上,再用干冰冻15 min。OCT包埋可有助于组织紧贴于冻头。
3.切片,并将切下的组织片转移至含有TCS的组织培养板的孔内(24或96孔板),此切片可长期储存于—20”C冰箱内
十、细胞培养板的包被


十一、Perodl密度梯变离心法纯化干细胞
![通过P ercoll密度梯度离心,可以去除污染的组织碎片和其他细胞,从而使干细胞得 以部分纯化。 1•按9 份 Perooll, 1 份 10X:PBS的 比 例 ( V/V),稀释Percoll储存液。 2 ■为获得不连续的PercoIl密度梯度,可在此基础上,制备实验所需的不同浓度 (50%、 4 0 % 、 3 0 % 、 2 0 % 和 1 0 % ) 的 Perooll梯度液,并将来源于酶消化和过滤后的细胞 悬液铺于 Perooll 梯 度 上 (Marie etal. 1997)。 3 . 室温条件下,离屯、 (400私, 20~30m in)。 4 . 收集分布于40% ~ 5 0 % P_ U梯度层之间的细胞。 5 . 将细胞用冷的PBS (含抗生素和两性霉素B) 稀释2〜5倍后离心(1〇〇〇 茗, 3min), 重复3 次。由于细胞沉淀较少,所以每次离心后,不应把上清液全部吸弃,而应留Iml左 右液体,以避免细胞丢失。 6 . 用 Iml培养液重悬细胞,取样计数,并以 个细胞/75on2 培养瓶]的密度接种细胞,如获得的细胞数较少,可将细胞接种于35mm 或 60mm的平皿中。 7 •通过Percoll连续密度梯度离心同样可分离纯化干细胞。将细胞与;Per00Il (1:1)混 合, i^ Li'后收集分布于上层( 髓憐脂层, myelin Iayor) 和 底 层 ( 红细胞层)之间的液体。 以后的操作可重复步骤5 和 6。](http://img.dxycdn.com/trademd/upload/userfiles/image/2016/07/B1468481624541pyuth2q43mpng_small.jpg)
十二、神经干细胞的传代和再培养
单层细胞的培养

神经球的传代
1.收集含有神经球的培养液并转移至15 ml无菌离心管中,离心(1000次,3 min), 缓慢吸弃上清液,注意不要搅混细胞沉淀。
2.用Iml含有EGF的无血清 N2 培养液重悬细胞,并用中等口径的巴氏吸管轻轻吹打10~20次,使其成为单细胞悬液。
3.将细胞接种于未经包被的培养皿内或冷冻于液氮中长期保存。
细胞冷冻
1.将细胞重悬于含1 〇%DMSO和合适生长因子的无血清N2培养液中。
2.用吸管轻轻吹打,使细胞均匀,然后以lml/管分装于无菌的冷冻管内。
3.将冷冻管直立放入泡沫小盒内,并置于一701〇冰箱,使细胞在冷冻过程中缓慶降温。
4.第二天,将细胞冷冻管移入液氮容器内。
冷冻细胞的复苏和培养
1.从液氮中取出冷冻细胞,立即投入37TC7JC浴中并不时摇动,令其尽快融化。
2.用DMEM:F12培养液稀释此融化细胞,并转移至15 ml无菌离心管中离心(lOOOg,3 min),吸弃上清液。
3.用培养液洗细胞1 次,然后将细胞重悬于Iml无血清N2培养液中,用中等口径巴氏吸管轻轻吸打,使其成为单细胞悬液。
4.将大鼠来源的细胞接种于经PORN/Laminin包被的培养皿内;将小鼠来源的细胞接种于未经包被的培养皿内,采用含合适生长因子的无血清N2培养液。
克隆细胞的传代(璋脂糖/胰酶法)
1.从培养皿中挑选相对较大(>1〇〇个细胞/克隆)且分离较好的克隆,并在培养皿背面做好标记。
2.于微波炉内融化 3% 的琼脂糖溶液(agarose; 用 PBS 配制),待冷却至 45~50℃时,将 1ml 琼脂糖溶液与 2 mlATV-胰酶混匀,于 37℃条件下保温。
3.吸弃培养皿内的培养液,立即加入琼脂糖/胰酶混合液,轻轻摇动平皿,使液体流遍细胞表面,待凝固 2~3 min。
4.用无菌巴氏吸管沿各细胞克隆四周轻轻割下,挑出粘附有克隆细胞的琼脂糖胶,逐一移至 24 孔板的各孔中(孔内含有添加了 50% 条件培养液和 G418 的无血清N2培养液),然后,在原先细胞克隆的生长处,用少量(IOOul) 培养液洗 2 次,洗液一并移入 24 板的相应孔内。
5.每隔 3~4d 换液 1次,直到培养的细胞达到所需的汇合度,准备传代。
十三、条件培养液的制备
1.从高细胞密度的培养液中(至少培养 24 h 以上)收集条件培养液。
2.离心(1 000 g,5 min),分装成小份后冻存。为了防止残留细胞的污染,也可将条件培养液过滤。
十四、脑切片的分析
移植的干细胞在体内的特性及其命运可通过免疫细胞化学染色、原位杂交和电镜观^等进行分析,这里仅介绍免疫细胞化学染色的方法。.
1.为检测 BrdU, 将 CNS来源的组织切片漂于 5%formamide/2XSSC 液中进行预处理(65T:,2 h), 然后,将切片置于 2mol/LHa 中,于 37℃ 孵育 30 min。
2.预处理过的切片分析采用上述的免疫细胞化学染色法,并可通过激光共聚焦或^光显微镜观察其特异性标志蛋白的表达,BrdU 和特异性标志蛋白的共定位将用以确定移植细胞在体内的命运。
脑切片的细胞数可通过无偏倚立体学法(unbiasedstereology) 来确定,该方法曾有过详细介绍(Sterio1984;Petersonetal.1994;WestandSIomianka1998),这里仅作简要介绍。
1.将包含研究对象的组织进行系列切片,以相同的、随机的方法抽取样本。依视觉解剖器原则(opticaldissectorprinciple) 存在于组织中的所有细胞被抽样计数的机会应该均等。
2.根据视觉解剖器的抽样原则,在三维无偏倚计数网格中直接计数细胞。
3.通过视觉分段步骤(theopiticalfractionatorprocedure) 可直接从已知的样本计算总体细胞数,或可将由视觉解剖计数中所获的密度值与由 Cavalieri 步骤(Nv-VRef 步骤)所估算的整个结构的体积相结合而得知。
来源:丁香实验