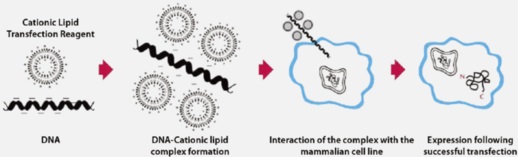
材料与仪器
步骤
材料
缓冲液与溶液
贮存液、缓冲液与试剂的成分见附录 1。
稀释贮存液至所需浓度。
DMSO(30%)/含血清培养基
第 3 步使用 DMSO 前,将高效液相(HPLC) 纯或组织培养纯的 DMSO 加入含血清的细胞生长培养基中至终浓度 30%(V/V)。
Giemsa 染液(10%m/V)
使用前用磷酸缓冲盐或水配制,用 Whatman1 号滤纸过滤。
甲醇
Polybrene(10 mg/ml)
用水溶解 Polybrene(Aldrich) 至终浓度 10 mg/ml,用 0.22 mm 的滤器过滤除菌, 分装成小等份(0.25 ml),-20°C 保存备用,使用后丢弃。
丁酸钠(500 mmd/L)(备选)
在化学通风橱内,用 10mol/LNaOH 调节丁酸钠贮存液至 pH7.0。用 0.22um 的滤器过滤除菌,分装成 1ml 的等份,-20°C 保存。
核酸与寡核苷酸
要转染的 DNA,如质粒 DNA, 用水溶解至 1ug/ul。
培养基
最低基础培养基(MEM)-α(含 10% 胎牛血清的培养基,不含血清的培养基与选择性培养基)
特殊设备
组织培养皿(90 mm)
此方法适用于用 90 mm 培养皿培养细胞。如果使用其他多孔板、细胞瓶或其他直径的培养皿,按比例改变细胞浓度与试剂的用量。见表 16-3。
附加试剂
本方案第 6 步可能需要第 17 章方案 7 或方案 1 辅助方案所列试剂。
细胞与组织
指数生长的哺乳动物细胞
方法
1. 通过胰酶消化收集细胞(如 CHO 细胞),将每 5X105 细胞用 10 ml 含 10% 胎牛血清的 MEM-α培养基平铺于 90 mm 组织培养皿上。于含 5%~7%CO2 的 37°C 温箱培养细胞 18~20 h。
2. 用 3 ml 温热的含血清、DNA(5ng~40ug,无载体 DNA)、30ug Polybrene 的培养基替换原来的培养基。先将 DNA 与培养基混勾再加入 10 mg/ml Polybrene。细胞继续孵育 6~16 h。孵育早期阶段每 90 min 摇动培养皿一次,使细胞均匀接触 DNA Polybrene 混合物。
3. 吸去含 DNA-Polybrene 的培养基,加 5 ml 含 30%DMSO、含血清的培养基。轻轻摇动培养基,使细胞均匀接触溶液,培养皿置于孵箱中。
4. 孵育 4 min 后,从孵箱中取出培养皿,快速吸去 DMSO 培养基。用无血清的温热培养基洗涤细胞一至两次,加入含 10% 胎牛血清的完全培养基。如果需要丁酸钠促进转染,则继续步骤 5, 如果不需要,则在含 5%~7%CO2 的 37°C 温箱培养细胞 48 h。然后直接进行步骤 6(检测瞬时转染)或 7(稳定转染)。
DMSO 处理的细胞易于脱落,因此在弃去含溶剂的培养基及加入新培养基时应尽可能动作轻柔,比如,每次换液时沿着培养皿侧壁吸培养基。
5.(选作)为了促进 DMSO 与 Polybrene 处理过的细胞的转染:
a. 在培养基中加入 500 mmol/L 丁酸钠至终浓度 2.5~10 mmol/L。
丁酸钠的确切浓度要根据细胞类型而定。
b. 在含 5%~7%CO2 的 37°C 温箱培养细胞 20~24 h。
c. 弃去含丁酸钠的培养基,换成含 10% 胎牛血清的无丁酸培养基。细胞于孵箱内培养。
丁酸钠可促进 DMSO 处理过的细胞的瞬时(不能持久)表达某种重组质粒(Aubin et al.1997),尤其是在猿与人细狍中的带 SV40 早期启动子/增强子的质粒(Gorman et al.1983a)。
6. 如果要稳定转染细胞,直接进行第 7 步。如果要瞬时表达则按照下述方法在转染 1~2 天后检测细胞:
• 如果使用的是表达 E.coli.β-半乳糖苷酶的质粒 DNA 按第 17 章方案 7 所列操作步骤检测细胞裂解液中酶活性。或者按照本章方案 1 的附加方案中介绍的组化染色方法操作。
• 如果使用绿色荧光蛋白表达载体,在 450~490nm 光照下用显微镜检测细胞。
• 如果表达的是其他基因产物,则通过体内代谢标志物进行放射免疫、免疫印渍、免疫沉淀或測定细胞提取物的酶活性来分析新合成蛋白。
为减小不同培养皿之间转染效率的差异,最好(1)用每一构建体转染数个培养皿;(2)孵育 24 h 后用胰酶消化细胞;(3)将细胞汇集起来;以及(4)重铺细胞于数个培养皿上。
7. 分离稳定转染体:用非选择性培养基培养细胞 48 h[使转移的基因表达(第 4 步)] 后,胰酶消化细胞或用适当的选择性培养基重铺细胞,也可以直接将选择性试剂加入培养基中。每 2~4 天换液一次,持续 2~3 周,目的是去除死细胞残骸,并允许抗性细胞克隆生长。
8. 此后,克隆,繁殖独立克隆以用于检测 [方法见 Jakoby and Pastan 1979 或 Spector et al.1998b(《细胞实验手册》的第 86 章)]
用预冷的甲醇固定细胞 15 min,然后室温下用 10%Giemsa 染色 15 min, 流水冲洗, 这样可以记录细胞克隆数目。Giemsa 染液应使用前用磷酸盐缓冲液或水新鲜配制,用 Whatman 1 号滤纸过滤。
缓冲液与溶液
贮存液、缓冲液与试剂的成分见附录 1。
稀释贮存液至所需浓度。
DMSO(30%)/含血清培养基
第 3 步使用 DMSO 前,将高效液相(HPLC) 纯或组织培养纯的 DMSO 加入含血清的细胞生长培养基中至终浓度 30%(V/V)。
Giemsa 染液(10%m/V)
使用前用磷酸缓冲盐或水配制,用 Whatman1 号滤纸过滤。
甲醇
Polybrene(10 mg/ml)
用水溶解 Polybrene(Aldrich) 至终浓度 10 mg/ml,用 0.22 mm 的滤器过滤除菌, 分装成小等份(0.25 ml),-20°C 保存备用,使用后丢弃。
丁酸钠(500 mmd/L)(备选)
在化学通风橱内,用 10mol/LNaOH 调节丁酸钠贮存液至 pH7.0。用 0.22um 的滤器过滤除菌,分装成 1ml 的等份,-20°C 保存。
核酸与寡核苷酸
要转染的 DNA,如质粒 DNA, 用水溶解至 1ug/ul。
培养基
最低基础培养基(MEM)-α(含 10% 胎牛血清的培养基,不含血清的培养基与选择性培养基)
特殊设备
组织培养皿(90 mm)
此方法适用于用 90 mm 培养皿培养细胞。如果使用其他多孔板、细胞瓶或其他直径的培养皿,按比例改变细胞浓度与试剂的用量。见表 16-3。
附加试剂
本方案第 6 步可能需要第 17 章方案 7 或方案 1 辅助方案所列试剂。
细胞与组织
指数生长的哺乳动物细胞
方法
1. 通过胰酶消化收集细胞(如 CHO 细胞),将每 5X105 细胞用 10 ml 含 10% 胎牛血清的 MEM-α培养基平铺于 90 mm 组织培养皿上。于含 5%~7%CO2 的 37°C 温箱培养细胞 18~20 h。
2. 用 3 ml 温热的含血清、DNA(5ng~40ug,无载体 DNA)、30ug Polybrene 的培养基替换原来的培养基。先将 DNA 与培养基混勾再加入 10 mg/ml Polybrene。细胞继续孵育 6~16 h。孵育早期阶段每 90 min 摇动培养皿一次,使细胞均匀接触 DNA Polybrene 混合物。
3. 吸去含 DNA-Polybrene 的培养基,加 5 ml 含 30%DMSO、含血清的培养基。轻轻摇动培养基,使细胞均匀接触溶液,培养皿置于孵箱中。
4. 孵育 4 min 后,从孵箱中取出培养皿,快速吸去 DMSO 培养基。用无血清的温热培养基洗涤细胞一至两次,加入含 10% 胎牛血清的完全培养基。如果需要丁酸钠促进转染,则继续步骤 5, 如果不需要,则在含 5%~7%CO2 的 37°C 温箱培养细胞 48 h。然后直接进行步骤 6(检测瞬时转染)或 7(稳定转染)。
DMSO 处理的细胞易于脱落,因此在弃去含溶剂的培养基及加入新培养基时应尽可能动作轻柔,比如,每次换液时沿着培养皿侧壁吸培养基。
5.(选作)为了促进 DMSO 与 Polybrene 处理过的细胞的转染:
a. 在培养基中加入 500 mmol/L 丁酸钠至终浓度 2.5~10 mmol/L。
丁酸钠的确切浓度要根据细胞类型而定。
b. 在含 5%~7%CO2 的 37°C 温箱培养细胞 20~24 h。
c. 弃去含丁酸钠的培养基,换成含 10% 胎牛血清的无丁酸培养基。细胞于孵箱内培养。
丁酸钠可促进 DMSO 处理过的细胞的瞬时(不能持久)表达某种重组质粒(Aubin et al.1997),尤其是在猿与人细狍中的带 SV40 早期启动子/增强子的质粒(Gorman et al.1983a)。
6. 如果要稳定转染细胞,直接进行第 7 步。如果要瞬时表达则按照下述方法在转染 1~2 天后检测细胞:
• 如果使用的是表达 E.coli.β-半乳糖苷酶的质粒 DNA 按第 17 章方案 7 所列操作步骤检测细胞裂解液中酶活性。或者按照本章方案 1 的附加方案中介绍的组化染色方法操作。
• 如果使用绿色荧光蛋白表达载体,在 450~490nm 光照下用显微镜检测细胞。
• 如果表达的是其他基因产物,则通过体内代谢标志物进行放射免疫、免疫印渍、免疫沉淀或測定细胞提取物的酶活性来分析新合成蛋白。
为减小不同培养皿之间转染效率的差异,最好(1)用每一构建体转染数个培养皿;(2)孵育 24 h 后用胰酶消化细胞;(3)将细胞汇集起来;以及(4)重铺细胞于数个培养皿上。
7. 分离稳定转染体:用非选择性培养基培养细胞 48 h[使转移的基因表达(第 4 步)] 后,胰酶消化细胞或用适当的选择性培养基重铺细胞,也可以直接将选择性试剂加入培养基中。每 2~4 天换液一次,持续 2~3 周,目的是去除死细胞残骸,并允许抗性细胞克隆生长。
8. 此后,克隆,繁殖独立克隆以用于检测 [方法见 Jakoby and Pastan 1979 或 Spector et al.1998b(《细胞实验手册》的第 86 章)]
用预冷的甲醇固定细胞 15 min,然后室温下用 10%Giemsa 染色 15 min, 流水冲洗, 这样可以记录细胞克隆数目。Giemsa 染液应使用前用磷酸盐缓冲液或水新鲜配制,用 Whatman 1 号滤纸过滤。
来源:丁香实验