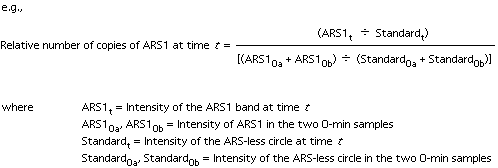Anatomy of a Comparative Gene Expression Study
互联网
Anatomy of a Comparative Gene Expression Study
Disclaimer: I'm a computer scientist, not a medical doctor. If you're interested in taking advantage of experimental diagnostic and therapeutic technologies, ask your doctor or visit, e.g., NCI's CancerNet web site .
DNA microarrays are perfectly suited for comparing gene expression in different populations of cells. The hows and whys of such an experiment provide insight into the power of microarrays, their limitations, and the kinds of biological questions which they can help to answer.
The illustration below shows the steps that make up a comparative cDNA hybridization experiment. Click on any of the steps in the image to jump to an explanation of how and why it is performed. Biotechnology terms which are not explained on this page are linked to a glossary, so only a minimal knowledge of modern biology is required. Enjoy!
If your browser can display Flash animations, Professor A. Malcolm Campbell of Davidson College has produced an animated description of comparative hybridization . Requests to use the animation should be directed to Dr. Campbell at macampbell@davidson.edu .
The major steps of a comparative cDNA hybridization experiment are
1. Choosing Cell Populations
The goal of comparative cDNA hybridization is to compare gene transcription in two or more different kinds of cells. We will describe some experiments of particular interest; however, the possibilities for informative comparative transcription studies are limited only by the investigator's imagination.
Tissue-specific Genes
Cells from two different tissues (say, cardiac muscle and prostate epithelium) are specialized for performing different functions in an organism. Although we can recognize cells from different tissues by their phenotypes , it is not known just what makes one cell function as smooth muscle, another as a neuron, and still another as prostate. Ultimately, a cell's role is determined by the proteins it produces, which in turn depend on its expressed genes. Comparative hybridization experiments can reveal genes which are preferentially expressed in specific tissues. Some of these genes implement the behaviors that distinguish the cell's tissue type, while other controlling genes make sure that the cell only performs the functions for its type.
Regulatory Gene Defects in Cancer
Genetic disease is often caused by genes which are inappropriately transcribed -- either too much or too little -- or which are missing altogether. Such defects are especially common in cancers, which can occur when regulatory genes are deleted, inactivated, or become constitutively active . Unlike some genetic diseases (e.g. cystic fibrosis) in which a single defective gene is always responsible, cancers which appear clinically similar can be genetically heterogeneous . For example, prostate cancer (prostatic adenocarcinoma) may be caused by several different, independent regulatory gene defects even in a single patient. In a group of prostate cancer patients, every one may have a different set of missing or damaged genes, with differing implications for prognosis and treatment of the disease.
Comparative hybridization can serve two purposes in studying cancer: it can pinpoint the transcription differences responsible for the change from normal to cancerous cells, and it can distinguish different patterns of abnormal transcription in heterogeneous cancers. Understanding the diverse basis of a cancer is crucial for inventing therapies targeted to the different varieties of the disease, so that each patient receives the most appropriate and effective treatment.
Cancers are common examples of genetically heterogeneous diseases, but they are by no means the only ones. Diabetes, heart disease, and multiple sclerosis are among the diseases for which genetic risk factors are known to be heterogeneous.
Cellular Responses to the Environment
How does a cell adapt to changes in its environment? Cells survive in the face of changes in temperature and pH, changing nutrient availability, and the presence of environmental toxins and ionizing radiation. Usually, a change in environment requires that expression of some genes be turned up or down so that the organism can respond appropriately. For example, common yeast has been extensively studied to understand how it switches between metabolizing sugars into ethanol and ethanol, in turn, into acetic acid (this is why wine with active yeast eventually becomes vinegar). The move from one metabolic state to the other, called diauxic shift , involves shutting down genes for processing sugars and activating others for processing ethanol, as well as a general stress response due to the greater difficulty of deriving energy from ethanol.
Comparative hybridization experiments can point out genes whose transcription changes in response to an environmental stimulus. In the simplest experiment, a population of cells is subjected to the stimulus and allowed to reach a steady state of transcription. Transcription levels in the altered cells can then be compared to those in a control population. A more informative experiment subjects cells to a change, then takes samples of the cell population at successive points in time. In this way, the experimenter can watch as the gene transcription patterns change from the old to the new steady state. Temporal studies can identify not only genes whose transcription changes but also the order of the changes, providing evidence about which genes control the response directly and which are only indirectly affected by it.
Environmental changes of interest also include the introduction of signaling molecules, such as hormones, interleukins, and interferons, as well as the actions of drugs. All these molecules stimulate a change in a cell's behavior (including possibly its death). While some of the changes may be mediated purely at the protein level, others require new transcription which can be detected by comparative hybridization.
Cell Cycle Variations
Even in a stable environment, cells undergo DNA replication, mitosis , and eventually death. These activities require quite different gene products, such as DNA polymerases for genome replication or microtubule spindle proteins for mitosis. A cell's genes encode the "programs" for these activities, and gene transcription is required to execute those programs. Comparative hybridization can be used to distingish genes that are expressed at different times in the cell cycle. In this way, the pathways responsible for controlling basic life processes can be uncovered.
2. mRNA Extraction and Reverse Transcription
Genes which code for protein are transcribed into messenger RNA's (mRNA's) in the cell nucleus. The mRNA's in turn are translated into proteins by ribosomes in the cytoplasm. The transcription level of a gene is taken to be the amount of its corresponding mRNA present in the cell. Comparative hybridization experiments compare the amounts of many different mRNA's in two cell populations.
To prepare mRNA for use in a microarray assay, it must be purified from total cellular contents. mRNA accounts for only about 3% of all RNA in a cell [1] , so isolating it in sufficient quantity for an experiment (1-2 micrograms) can be a challenge. Common mRNA isolation methods take advantage of the fact that most mRNA's have a poly-adenine (poly(A)) tail. These poly(A)+ mRNA's can be purified by capturing them using complementary oligodeoxythymidine (oligo(dT)) molecules bound to a solid support, such as a chromatographic column or a collection of magnetic beads.
Captured mRNA's are still difficult to work with because they are prone to being destroyed. The environment is full of RNA-digesting enzymes (there are some on your fingers, your keyboard, your mouse, and every other exposed surface around you right now), so free RNA is quickly degraded. To prevent the experimental samples from being lost, they are reverse-transcribed back into more stable DNA form. The products of this reaction are called complementary DNA's (cDNA's) because their sequences are the complements of the original mRNA sequences. The reverse transcription reaction usually starts from the poly(A) tail of the mRNA and moves toward its head; such a reaction is called oligo(dT)-primed .
A problem with cDNA production is that not all mRNA's are reverse-transcribed with the same efficiency. This fact leads to reverse transcription bias , which can change the relative amounts of different cDNA's measured by the microarray assay. Reverse transcription bias is not a problem when comparing the same mRNA across two cell populations unless it causes the mRNA not to be transcribed at all. However, the bias does prohibit quantitative comparison between different mRNA's on one array. Another problem caused by bias is that some mRNA's may be reverse-transcribed for only part of their lengths, making them less likely to bind and stay bound to their complements on the array. One way of getting around this problem is to prime reverse transcription from random starting positions on the mRNA's, rather than always starting from their tails. This method can reduce bias, but it also makes useless cDNA from any remaining ribosomal and transfer RNA's in the sample.
3. Fluorescent labeling of cDNA's
In order to detect cDNA's bound to the microarray, we must label them with a reporter molecule that identifies their presence. The reporters currently used in comparative hybridization to microarrays are fluorescent dyes (fluors), represented by the red and green circles attached to the cDNA's in the diagram [2] . A differently-colored fluor is used for each sample so that we can tell the two samples apart on the array. The labeled cDNA samples are called probes because they are used to probe the collection of spots on the array.
The colors of the fluors in the diagram are just for illusrtration. The actual fluors do not show their colors unless stimulated with a specific frequency of light by a laser. Even then, the colors are not directly observed; rather, the wavelength of the emitted light is used to tune a detector which measures the fluorescence. The choice of red and green colors is reminiscent of the emission wavelengths of commonly-used fluors, such as rhodamine and fluorescein or Cy3 and Cy5.
The number of fluor molecules which label each cDNA depends on its length and possibly its sequence composition, both of which are often unknown. This is one more reason that fluorescent intensities for different cDNA's cannot be quantitatively compared. However, identical cDNA's from the two probes are still comparable as long as the same number of label molecules are added to the same DNA sequence in each probe.
To equalize the total concentrations of the two cDNA probes before applying them to the array, the probe solutions are diluted to have the same overall fluorescent intensity. This procedure makes two possibly unjustified assumptions: first, that the total amount of mRNA in each cell type being tested is identical; and second, that each fluor emits the same amount of light relative to its concentration. The second assumption can be eliminated by suitable calibration, but the first one is difficult to check. It may therefore be that cells with more abundant mRNA are made into a probe with artifically low mRNA concentrations.
4. Hybridization to a DNA Microarray
The two cDNA probes are tested by hybridizing them to a DNA microarray. The array holds hundreds or thousands of spots, each of which contains a different DNA sequence. If a probe contains a cDNA whose sequence is complementary to the DNA on a given spot, that cDNA will hybridize to the spot, where it will be detectable by its fluorescence. In this way, every spot on an array is an independent assay for the presence of a different cDNA. There is enough DNA on each spot that both probes can hybridize to it at once without interference.
Microarrays are made from a collection of purified DNA's. A drop of each type of DNA in solution is placed onto a specially-prepared glass microscope slide by an arraying machine . The arraying machine can quickly produce a regular grid of thousands of spots in a square about 2 cm on a side, small enough to fit under a standard slide coverslip. The DNA in the spots is bonded to the glass to keep it from washing off during the hybridization reaction.
The choice of DNA's to be used in the spots on a microarray determines which genes can be detected in a comparative hybridization assay. For organisms whose genomes have been completely sequenced, including several bacteria and the the yeast Saccharomyces cerevisciae , it is possible to array genomic DNA from every known gene or suspected open reading frame (ORF) in the organism. Each gene or ORF is amplified from total genomic DNA by PCR , producing enough DNA to make unlimited numbers of arrays. The Pat Brown Lab at Stanford University has arrayed all known or suspected genes of S. cerevisciae (roughly 6100) on a single microarray .
Because the human genome has not been completely sequenced, we cannot yet produce a comprehensive array for all its genes. Moreover, the number of human genes has been estimated at somewhere between 10,000 and 100,000, so several arrays will probably be required to hold them all. Despite these limitations, several strategies can be used today to make arrays for studying human genes. We do know the location and sequence of quite a few human genes now, so the same method used to array yeast genes will produce at least a partial human genome array. There are two other ways to produce arrayable DNA even for unknown genes: amplify clone inserts from human cDNA libraries , or synthesize oligonucleotides directly from known expressed sequence information such as EST's . While neither of these methods will produce DNA's for every human gene, both can yield enough different expressed sequences to make substantial arrays. Both types of DNA have been used before in array-like applications: cDNA libraries were used for comparative hybridization before the advent of fluorescent microarrays, while oligonucleotide arrays are available commercially today from Affymetrix Corporation for rapid resequencing of a few genes important to AIDS and some cancers.
5. Scanning the Hybridized Array
Once the cDNA probes have been hybridized to the array and any loose probe has been washed off, the array must be scanned to determine how much of each probe is bound to each spot. The probes are tagged with fluorescent reporter molecules which emit detectable light when stimulated by a laser. The emitted light is captured by a detector, either a charge-coupled device (CCD) or a confocal microscope , which records its intensity. Spots with more bound probe will have more reporters and will therefore fluoresce more intensely.
Each of the two fluorescent reporters (fluors) used has a characteristic excitation wavelength ; only light of this wavelength will cause the molecule to fluoresce. The emitted light has a characteristic emission wavelength which is different from the excitation wavelength. The detector for the emitted fluorescence from the array is sensitive to the emission wavelength but filters out the excitation wavelength; in this way, the fluorescent light of interest can be separated from the laser light scattered off the slide.
A good pair of fluors for a comparative hybridization experiment should have very different emission or excitation wavelengths. If the emission wavelengths are different, light emitted from the two fluors can be selectively filtered to measure the amount emitted by each fluor separately. If the excitation wavelengths are different, the two fluors can be stimulated and scanned one at a time. If one of these conditions is not met, the scanned intensities can be contaminated by crosstalk between the two fluorescent channels.
Although the purpose of the scanner is to pick up light emitted by probe cDNA's bound to their complementary spots, it also records light from a few molecules that hybridized either to the wrong spot or nonspecifically to the glass slide. This extra light becomes the background of the scanned array image. One advantage of current microarray technology over its predecessors is that the background is extremely low; consequently, the signal-to-noise ratio of the scanned data can be quite high.
6. Interpreting the Scanned Image
The end product of a comparative hybridization experiment is a scanned array image. A small piece of such an image is shown above. The measured intensities from the two fluorescent reporters have been false-colored red and green and overlaid. Yellow spots have roughly equal amounts of bound cDNA from each cell population and so have equal intensity in the red and green channels (red + green = yellow). Spots whose mRNA's are present at a higher level in one or the other cell population show up as predominantly red or green.
The intensities provided by the array image can be quantified by measuring the average or integrated intensities of the spots. The ratio of fluorescent intensities for a spot is interpreted as the ratio of concentrations for its corresponding mRNA in the two cell populations. Schena et al (1996) have demonstrated the ability to detect quantitative changes of as little as a factor of two, with reasonable agreement between expression ratios measured on the array and ratios measured by an alternate form of RNA blotting.
Interpreting the data from a microarray experiment can be challenging. Quantitation of the intensities on each spot is subject to noise from irregular spots, dust on the slide, and nonspecific hybridization. Deciding the intensity threshold between spots and background can be difficult, especially when the spots fade gradually around their edges. Detection efficiency might not be uniform across the slide, leading to excessive red intensity on one side of the array and excessive green on the other side. Even after overcoming detection and calibration problems, the measured intensities for each spot only represent the ratio of cDNA's in each cell population. Low levels of cDNA due to reverse transcription bias, sample loss, or an inherently rare mRNA can cause large uncertainties in these ratios.
Numerous software packages, both free and commercial, exist for quantitating microarray data. I have developed one such program, Dapple , to address some of the abovementioned image quality issues, in the hope that its methods might be integrated into other array quantitation systems.
Typically, the interpreted array data will highlight a relatively small number of spots representing very differentially-expressed mRNA's whose genes deserve further investigation. Alternatively, the overall pattern of expression can be used as a "fingerprint" to characterize specific cell types (e.g. different classes of tumors), even if not all the differentially-expressed genes on an array have been identified.
Notes
[1] The rest is ribosomal RNA (rRNA) and transfer RNA (tRNA).
[2] Predecessors to current microarray technology added radioactive phosphorus to the cDNA molecules, so that hybridized cDNA's would form spots on X-ray film (or more sensitive phosphorimaging devices). This labeling technology is inadequate for comparative hybridization to the same microarray, since we have to distinguish the two different samples from each other.