DNA Extraction
互联网
This is a modification of a salting out procedure as described by Miller et al., (1988), evaluated at the DNA Laboratory, Medical School, Malta. When analysed by spectrophotometry >95% of extracted genomic DNA gave a purity ratio of DNA to proteins in the range of 1.7 - 1.9 and a concentration of 100ng/µl.
In the following protocol DNA was extracted from peripheral blood leucocytes using 3mls of whole blood. It was observed that on prolonged storage of whole blood at -20 and -80ºC , DNA yield decreased considerably probably due to degeneration of the white blood cells. Volumes of buffers and reagents used may be adjusted according to volume of whole blood used.
PROCEDURE:
1.Whole blood was collected in disodium EDTA containers and stored at -20ºC and a number of samples were also stored at -80ºC. To facilitate haemolysis of RBCs it is recommended to store a fresh sample for a few hours in a freezer as freezing destroys the red cells.
2.After thawing, 3mls of whole blood are transferred to a sterile conical centrifuge tube (15ml volume) to which 9mls of 1 x erythrocyte lysing buffer (0.155M NH 4 Cl; 10mM KHCO 3 ; 0.1mM Na2 EDTA; pH 7.4) must be added.
3.The solution is left for 10 minutes at RT with occasional mixing by inversion followed by centrifugation for 5 minutes at 4000 rpm.
4.After centrifugation the supernatant is discarded and a white pellet will be observed at the bottom of the tube. This pellet must be washed for at least 3 times by adding 3mls of erythrocyte lysing buffer and repeating steps 3 and 4. It is important to breakdown the pellet and rinse it well in erythrocyte lysing buffer in order to clean the white blood cells from remaining heme.
5.1.5mls of SE buffer (75mM NaCl; 25mM Na2 EDTA; pH 8.0) containing 100mg/ml of Proteinase K and 1% sodium dodecyl sulphate (SDS w/v), are added to the pellet. The tubes are then incubated at a temperature of 37 - 55ºC (optimal temp for Proteinase K activity) overnight in a water bath or incubator. During this step the white blood cells' membranes are denatured and DNA goes out in solution.
6.After the incubation, 1.5mls of SE buffer together with 750ml of 6M (saturated) NaCl are added to each tube, followed by the addition of 3.75mls chloroform.
7.The tubes are mixed vigorously (on a vortex) for about 20 sec with occasional mixing for at least 30min. Alternatively you can leave the tubes on a rotator for 1 hour.
8.The emulsion will then be centrifuged for 10 minutes at 2000 rpm with minimal breaking force. After centrifugation 2 phases are observed and care must be taken not to disturb the interphase. During this step DNA is extracted into the supernatant and proteins separated into the lower phase.
9.The upper aqueous phase (containing the DNA) is transferred into a clean and sterile conical centrifuge tube using a sterile Pasteur pipette, followed by the addition of an equal volume of isopropanol (double the volume of 100% ethanol can also be used)
10.DNA will be precipitated by gentle swirling of the tube and is observed visually as a white thread like strand.
11.Using a sterile spatula or loop transfer the DNA strand into a sterile microcentrifuge tube containing 1ml of 75% ethanol.
12.The DNA is then washed by inversion to clean it from any remaining salts and the tube centrifuged at 11000g for 4 minutes. The supernatant is discarded taking care not to discard the pellet. Repeat this step once more.
13.After discarding the supernatant the pellet is dried from excess ethanol either by using a vacuum centrifuge or by leaving the tubes open and inverted in an oven at around 50 - 65ºC for an hour.
14.The dried pellet is resuspended in TE buffer (1M Tris-HCl; 0.5M EDTA; pH 8.0) and left overnight on a rotator.
15.DNA concentration is determined either by agarose gel electrophoresis or spectrophotometry and adjusted to the desired concentration by adding more TE buffer. It is important to note that before adjusting and reading DNA concentrations one must obtain a homogeneous sample of DNA which is not quite easy acquired since DNA is very viscous. To adjust to lower concentration one must used other quantitation methods such as Picogreen (Molecular Probes) using spectrofluorometry as spectrophotometry is not accurate and sensitive at very low concentrations.
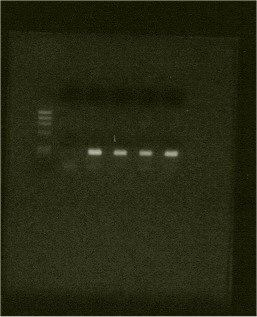
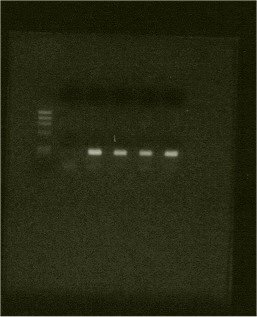
Fig. 1. Col1a1 Sp1 Fragment amplified by PCR using DNA extracted by the above method.
DNA STORAGE2
For routine storage the best condition is at +4 degrees Celsius in TE buffer (pH 8.0). Highly purified DNA can be stored for longer time.
Dried DNA pellets can be stored at -20 degrees Celsius for up to 6 months.
DNA precipitated in ethanol can be stored indefinitely at -20 degrees Celsius. For long term storage -80 degrees Celsius is recommended.
REFERENCE
1. Miller SA, Dykes DD, Polesky HF 1988 A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16: 1215.
2. Green et al. 1998 Genome Analysis: A Laboratory Manual. Cold Spring Harbor Laboratory Press.