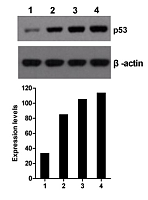
小RNA的Northern BLOT
互联网
- 相关专题
- Northern Blotting技术专题
Prepare glass plates for gel:
Rinse plates with cold water, then clean with dishwashing liquid and warm water. Do not scratch plates. Rinse with hot tap water until all soap is removed. The water should form an even sheet over the plate. Rinse thoroughly with tap distilled water, then double distilled water. Rinse with 95% Ethanol and wipe dry with kimwipes, make sure all dust is gone.
Also wash spacers and comb with soap and water. Rinse with distilled water; double distilled water and ethanol.
Put 1.5 mm thick spacers on the 20 x 40 cm glass plate, put the notched glass plate on top of the spacers. Clamp the plates together on one side, top and bottom with large binder cliPS .
10% Denaturing Acrylamide gel, 100ml (for 20x40 cm glass plates 1.5mm thick)
Swirl and warm up to 42C to allow for rapid polymerization (Heating is optional and not suggested for first-timers )
Pour gel:
Add 678 μl (i.e. if using 100 mls of Urea/Acrylamide) 10% APS (Date:________)
and
67.8 μl TEMED (Lot#___________), swirl to mix and start pouring immediately
with a 50 ml pipette.
Hold the plates at approximately 45 degrees while pouring. Tap the glass to get rid off bubbles. Insert comb and slowly lower the plates down to almost horizontal (put two pipette tip boxes under gel). Pour more gel around comb, wait a few minutes and then try to position the gel horizontal. Leave to polymerize about 1 hour (this depends on the
temperature of the acrylamide, as well as how much temed and APS is added; again I suggest warming the acrylamide up to allow for rapid polymerization). If not running the gel immediately, cover with saran wrap to leave overnight at room temperature.
Pre-running the gel:
Carefully remove comb and spacer on bottom and rinse off any excess acrylamide and urea around comb with syringe. Make sure the wells look clean and no more loose pieces of gel or urea is left.
Stand the gel in the lower buffer reservoir so that the notched glass plate faces the top reservoir (make sure the seal is in it’s groove) and the metal plate in the back. Clamp the gel with the metal plate at the top reservoir on both sides. Make sure the clamp is putting pressure over the spacer between the glass plates and that the clamp is in the
middle of the gasket and not in the middle of the reservoir. The metal plate should not come in contact with the buffer.
Add 1x TBE buffer in both reservoirs. Rinse out the wells with 1x TBE buffer, using a syringe.
Pre run the gel at constant 65 mA until the gel reaches 50℃, about 1 hr. (Glue a temperature strip on the metal plate to monitor the temperature of the gel.)
Load and run the gel:
Mix sample with equal volume 95% Formamide (with BromoPhenol Blue and Xylene cyanol FF).
Heat at 70℃ for 2 min, move to ice.
Turn off power. Rinse out the wells with 1x TBE using a syringe and needle, make sure all urea is rinsed out, load samples.
Run gel at constant 40 mA for about 1 hr (BPB runs around 12nt and cyanol around 55nt.)
UV-Shadow and Semi-dry transfer:
Take out all buffer from top reservoir with syringe. Un-clamp gel.
Pry apart glass plates with spatula. Cut off upper right corner of gel. Place saran wrap on top of gel, flip over and remove other glass plate. (Be sure to wet the gel with a little TBE so it does not dry out and break!!)
UV shadow gel with TC plate and take a picture to ensure samples are evenly loaded (although will eventually want to reprobe blot with a loading control).
Put a piece of Whatman 3M paper (pre-wet with 1x TBE) on gel, put glass plate (or parafilm) on top and flip over.
Measure gel and cut a piece of Hybond N+ to size (Lot#________________,) use clean gloves and scissors. Prewet in 1x TBE, position Hybond on gel, roll out bubbles with plastic sterile pipette. Put 1 piece of pre-wet (1x TBE) Whatman 3M paper on top. Transfer gel/membrane/whaman paper sandwhich to semi-dry transfer apparatus.
Wipe off excess liquid around the edges of gel. Put lid on. Plug in the power supply and transfer at 250mA for 2 hours or overnight. Note: 300mA is the max on our power supply.
5’end-labeling of Probe:
1 μl 10x PNK buffer (fresh from NEB)
30 pmol probe DNA oligo
1.66 μl [γ -32P] ATP, 150 mCi/ml (= 250 μCi, 42 pmol) Vial#______________
1 μl T4 Polynucleotide Kinase, 10 U/μl Lot#______________
X μl Nuclease free H2O to 10 μl
37℃ at least 1 hr
After incubation, bring reaction volume up to 50 μl before cleaning up with spin column! If too little volume is used, it will become trapped in column.
Microspin G-25 column: Lot#______________
Resuspend the resin in the column by vortexing gently
Loosen the cap 1/4 turn and snap off bottom, place in 1.5 ml screwcap eppendorf tube
Pre spin column for 1 minute @ 735xg (start timer and fuge simultaneously).(Marathon 16K: 2900 rpm = 700xg, 3000rpm = 800xg)
Place column in new screwcap tube and slowly apply sample to center of angled surface of resin bed. Do not disturb the resin.
Spin column for 2 minutes @ 735xg. Discard column in radioactive waste.
Take out 1 μl of labeled probe in 0.5 ml eppendorf tube, put tube in scintillation vial and count the cpm. Count:_______________
UV Crosslink:
UV Crosslink the blot at 1200 x 100 μJoles after transfer.
Optional: Allow membrane to dry overnight. Some evidence suggests this may improve sensitivity.
Wet blot in 1xTBE, roll blot with transfer side in (upper right corner off) and put in hyb bottle. Try to get rid of all air bubbles between glass bottle and blot. Pour out any excess liquid.
Pre-Hyb: Ambion ULTRAhyb-oligo Lot#______________
Heat hyb buffer @ 65℃ briefly to dissolve any precipitated material, swirl bottle often.
Take out ~25 ml (1ml/10cm2) in 50 ml conical tube, preheat @ 37℃.
Add hyb solution to hyb bottle and pre-hyb for at least 5 min. Pre-hyb:___________
Hybridization:
Pour out ~10 ml of pre-hyb into conical tube(keep “old” tube used to pre warm the hyb soln at 37 degrees) , add probe and mix, return to hyb bottle
Hybridize O/N @37℃ (this incubation may be shortened to as little as 2 hours, but this should be optimized for each probe).
Start hyb @ 37℃:________________ Stop:___________ Hyb time:__________
Wash buffer, 2xSSC / 0.1%SDS:
Mix: 50 ml 20x SSC + 445 ml DEPC H2O + 5 ml 10% SDS
Pre heat 2 x 50 ml @ 37℃.
Wash: Sart time:______________
~undefined*Wash_times_can_be_shortened_if_necessary~3_I_have_done_as_little_as_5’, but may depend on your probe. If you dry your membranes overnight, wash times may need to be increased.
Pour out hyb buffer in radioactive liquid waste. Turn off temperature in hyb oven (set temp to 22℃).
Add 50 ml 37℃ wash buffer, put hyb bottle in hyb oven for 1 min rotating but temperature off.
Pour out into liquid waste, add 50 ml 37℃ wash buffer, rotate for 30 min in hyb oven (temp set @22℃). Temperature after 30 min:____________
Repeat washes with 2 x 50 ml RT wash buffer, 30 minutes each.
-Tip. If background is still very high (as determined by Geiger counter) may want to continue washing blot at 37C.
Take out blot with clean forceps into a tray with wash buffer. Drain off excess liquid and put blot in saran wrap. Squeeze out air bubbles and excess liquid. Tape edges. Put blot in film cassette, add positioning markers.
Place blot in intensifier screen (these are great if you own one!), add Kodak MS film (greater sensitivity with intensifier screen) and expose blot at -80C.
I usually do a 1-3 hour exposure to get an idea of how long I will need to expose. For really highly expressed miRNA s (i.e. miR-124, may only need 1 hour). For lowly expressed, may need several days. Alternatively, we also use a phosphorimager to image blots in lieu of film, which is preferred if you are planning to perform quantitation.
REAGENTS and SOLUTIONS
SequaGel (National Diagnostics cat# EC-833)
Temed (BioRad cat# 161-0800)
SafetyCoat: Baker cat# 4017-01
10% APS, Make a 10% w/v in DEPC treated H2O. Ammonium persulfate, Sigma A7460. Keep in refrigerator, make fresh every week.
10xTBE: (890 mM) 216g Trisbase Fisher BP 152-5 (MW 121.14)
(890 mM) 110g Boric acid Fisher (electroph.gr) BP 168-500 (MW 61.83)
(20 mM) 40 mL 0.5M EDTA pH 8.0
H2O to 2L, check pH w. strip (take out and drizzle ~500 ul on
pH strip, ~pH 8.5, filter sterilize 0.2um, Fill 3 one liter bottles, mark level,
autoclave and bring up to level with H2O after cooling to RT
Fluka Bromophenol Blue-Xylene Cyonol solid mixture cat no 18047, when reconstituted to 5 ml, ~ 0.5% each dye in Tris-borate-EDTA buffer pH 8.3. Add 1.39 ml DEPC H2O~1.8% dye. Mix 95 uL Formamide with 5 uL 1.8% Bromophenolblue/Xylene cyanolFF
Note: can use less dye.
Hybond N+: Amersham RPN303B
ULTRAhyb-Oligo Hybridization Buffer: Ambion cat # 8663, keep in refrigerator
T4 Polynucleotide Kinase: NEB M0201S
Gamma 32P ATP, 6000 Ci/mmol, 150 mCi/mL PerkinElmer (cat# NEG035C005MC)
MicroSpin G-25 columns: Amersham 27-5325-01 (cellculture)
Wash buffer: 2xSSC/0.1%SDS
50 mL 20x SSC + 447.5 mL autoclaved H2O + 2.5 ml 20% SDS
This protocol has been put together with the help of many people from the McManus lab and surrounding labs at UCSF! Thanks for all of your help.