Computational Analysis of ProteinDNA Interactions from ChIP-seq Data
互联网
416
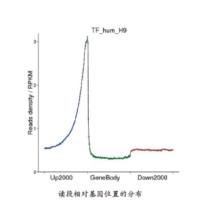
Chromatin immunoprecipitation experiments followed by ultra-high-throughput sequencing (ChIP-seq) is becoming the method of choice to identify transcription factor binding sites in prokaryotes and eukaryotes in vivo. Here, we review the computational steps that are necessary for analyzing the sequenced chromatin fragments, including mapping of short reads onto reference genomes, normalization of multiple conditions, detection of bona fide peaks or binding sites, annotation of sites, characterization of sequence-specific binding affinities, and relationships with biophysical models for protein–DNA interactions. The goal is that following the indicated steps will help the discovery of novel mechanisms underlying transcription regulation in a broad range of experimental systems.