DNA extraction from Mutation Detection Enhancement (MDE) Gel
互联网
Abstract: Single strand conformational polymorphism (SSCP)is the most widely used PCR-based methods for point mutation detection. The abnormal band found by SSCP analysis is normally verified using sequencing. There are many different procedures to recover DNA fragments from the silver stained polyacrylamide gels. However, most of those techniques are time consuming and their recoveries can be low for the Mutation Detection Enhancement (MDE) gel. This is a rapid and efficient method for recovering DNA fragments from MDE gel. In this procedure, the authors have suggested boiling in phenol/ chloroform/isoamylalcohol for elution of DNA from MDE gel that gives high yield of DNA without swelling of the gel. After precipitation of DNA by ethanol and DNA carrier such as glycogenat -70°C, the DNA pellet is rinsed with 75% ethanol to remove contaminations. The yield and quality of resulting DNA from the SSCP variant by this method is sufficient for PCR re-amplification, which can be followed by DNA sequencing to identify mutation. This method can be similarly used for DNA elution from polyacrylamide gels.
Detecting an unknown mutation may be compared to finding a needle in a haystack. The ability to find a mutation, which may alter as little as one billionth of the genome, clearly requires a very sensitive technique. Nowadays, that sensitivity is largely provided by PCR (5). Single-Strand Conformation Polymorphism (SSCP) is the most widely used DNA screening method. SSCP detects single-base sequence changes by abnormal electrophoretic migration of one or both single strands on a nondenaturing polyacrylamide gel (3). Recently some companies introduce a new gel matrix called Mutation Detection Enhancement (MDE), which has advantages over conventional polyacrylamide in resolving subtle mobility difference in DNA. The abnormal band found by SSCP is normally verified using sequencing. The most important part of sequencing reaction is to prepare sufficient and clean DNA template. To enrich mutant DNA prior to sequencing, aberrantly migrated bands were excised from the SSCP gel and then re-amplified by PCR, which can be subjected to direct DNA cycle sequencing. Many different procedures are now available to recover the DNA fragments from polyacrylamide gel (4, 6). However, those techniques can take a long time and recoveries can be low for MDE. We present a rapid, convenient and sensitive technique for recovering DNA fragments from MDE gel, stained with silver nitrate, particularly when aberrant bands have low concentration of DNA.
The protocol outlined in detail below has been used successfully to improve mutation detection in exon3 of the ¦Â-catenin (CTNNB1) gene in gastric adenocarcinoma by PCR-SSCP method. Following extraction of genomic DNA from paraffin embed allowfullscreen='true'ded tissues (7), 5 to 10µl of the crude extract were amplified in 25 µl reaction containing 10mM Tris-Hcl (pH 8.3), 50 mM KCl, 2 mM MgCl2, 50µM dNTP, 0.5µM each primer, and 0.6 unit of Taq DNA polymerase (Cinagen, Tehran, IRAN). The primers used were 3-A (5'-TAGTCACTGGCAACAGTCTT-3') and 3-B (5'-AAAATCCCTGTTCCCACTCATAC¨C3'); these amplified a 146-bp fragment of exon 3 of the CTNNB1 gene, encompassing the sequence for GSK-3¦Â phosphorylation. The PCR conditions consisted of 1 cycle at 95˚C for 5 min; 40 cycles at 95˚C for30 sec, 55˚C for 45 sec, and 72˚C for 75 sec; at a final extension cycle at 72 ˚C for 5 min. PCR reactions were performed in Gene Amp® PCR System 480 (Perkin-Elmer, Norwalk, CT, USA). DNA samples of normal subjects (blood donors) were included as well as negative control (with no DNA sample) in each reaction. Because most of the tumors were over 5 years old and extensive DNA degradation is a common event in old paraffin embed allowfullscreen='true'ded samples, 40 PCR cycles were necessary to create enough amplification products for SSCP and sequencing. Reaction products were separated in 3% agarose or 5% polyacrylamide gels and stained in ethidium bromide for visualization (1). The samples were diluted 1:9 in loading buffer (95% formamide, 10mM NaOH, 0.05% bromophenolblue, and 0.05% xylene cyanol), heated at 95˚C for 5 min, and quickly plunged into ice, then 5µl were loaded to (12 ¡Á 17 ¡Á 0.04 cm) 0.5X MDE (BMA, Rockland, ME, USA) gel containing 10% glycerol. Electrophoresis was carried out using 1X TBE (0.098 M Tris-Hcl, 0.098M boric acid, 10Mm EDTA) at room temperature with the power fixed at 200v for 5-6 h (8). After electrophoresis the gel was stained with silver nitrate to visualize DNA bands (2).
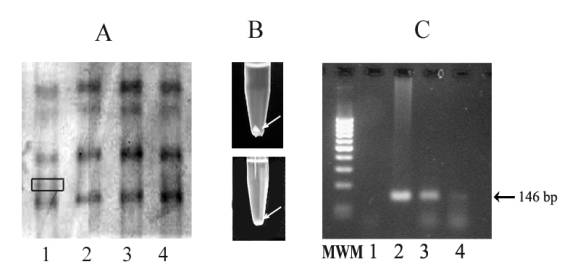
We managed to directly re-amplifying mutant DNA fragments after recovering from the gel. Extraction of DNA from MDE was preformed as follows: two SSCP variants containing the same concentration of DNA were removed from the MDE gel. DNA concentration has been measured with Kodak Digital Science 1D Image Analysis Software (Eastman Kodak, Rochester, NY, USA). One of them undergoes the conventional method (6) and the other gel slice was transferred into microcentrifuge tube and then completely crushed by a disposable pipette tip. In the next step, 100-200 µl volume of phenol/chloroform/isoamyl alcohol (25:54:1) was added to the microcentrifuge tube and incubated at room temperature for 30 min. The mixture was boiled at 100℃ for 15 minutes and centrifuged at 10,000-¡Á g for 1 min with model RM14 Rotor (Sorvall, Newtown, CT, USA). The supernatant was transferred to a new microcentrifuge tube and added 3-3.5 volumes of 100% ethanol, 1/10 volumes sodium acetate (pH 5.2) and 5-10µl glycogen (10 mg/ml) used as a carrier, followed by incubation at -70℃ for 30 min to precipitate DNA. After Centrifugation at 12000-¡Á g for 10 minutes, the supernatant was removed and the DNA pellet was rinsed with 100µl of 75% ethanol. This step is crucial, because precipitating DNA under this condition limits contamination and allows redissolution of the pellet at the desired concentration. DNA was again precipitated by centrifugation at 12000-¡Á g for 5 minutes. The pellet is completely air dried and dissolved in 10-20µl H 2 O overnight at 37℃. All reagents were purchased from sigma (Corning®; Sigma Chemical). The yield of DNA from a single band was usually too week to be visualized on agarose gel stained with ethidium bromide but 5µl of resulting DNA was sufficient for PCR reaction. This re-amplification was carried out under the same conditions as described above except that the reaction was completed in 35 cycles (Figure 1).
In conclusion, different methods have been developed for the recovering DNA from polyacrylamide gel. In the most of these protocols the gel pieces were soaked in water at different temperatures, but MDE gel swells after incubation in water and DNA elution from the gel obtains low quantity. Therefore, DNA is lost at the precipitation steps (Figure 1B). In this reason, DNA yield is not sufficient to re-amplify well by PCR (Figure 1C). Described method provides a convenient means of recovering DNA fragments in sufficient amounts and quality from MDE gel stained with silver nitrate in less than 2 hours.
Figure 1. Recovering DNA fragments from MDE gel after SSCP analysis. (A) SSCP analysis of exon 3 of the ¦Â-catenin gene. Lanes 2-5 represent individuals with a normal band and lane 1 is a suspected mutation carrier. Band with altered mobility that enclosed in box was eluted from the gel by standard method and described protocol. (B) Comparison of DNA precipitation after recovering aberrant migrated band from MDE gel by described method (upper microcentrifuge tube) and conventional method (lower microcentrifuge tube). Arrows show DNA precipitation. (C) 3% agarose stained with ethidium bromide, showing the results of re-amplification of single-strand DNA fragments obtained from MDE gel by conventional method (lane 3) and described method (lane 4). Genomic DNA prepared from whole blood was used as a positive control (lane 2). MWM and lane 1 denote the size marker (100-bp) and negative control (without DNA), respectively. The size of PCR fragment was 146-bp.
REFERENCES
1. Bian, Y.S., M.S. Osterheld, F.T. Bosman, C. Fontolliet and J. Benhatlar. 2000. Nuclear accumulation of ¦Â-catenin is a common and early event during neoplastic progression of Barrett esophagus. Am J Pathol. 114:583-590.
2.Chaubert, P., D. Bastita and J. Behattar. 1993. An improved method for rapid screening of DNA mutations by nonradioactive single-strand conformation polymorphism procedure. BioTechniques. 15:586.
3.Cotton, R.G.H. 1997. Mutation Detection, P.32-58. Oxford University Press, New York.
4.Etokeb, G.E. and A. Sparkland. 2000.Method for avoiding PCR-inhibiting contaminants when eluting DNA from polyacrylamide gels. BioTechniques. 29:694-696
5.Hawkins, J.R. 1997. Finding mutation, P.56-69. Oxford University Press, New York.
6.Sambrook, J., E.F. Fritsch and T. Maniatis. 1989. Molecular cloning: A Laboratory Manual, 2nd ed. CSH laboratory press, Cold Spring Harbor, NY.
7.Sepp, R., I. Szabo, H. Uda and H. Sakamoto. 1994. Rapid techniques for DNA extraction from routinely processed archival tissue for use in PCR. J Clin. Pathol. 47:318-323
8.Voeller, J.H., C.I. Truica and E.P.Gelmann. 1998. ¦Â-catenin mutations in human prostate cancer. Cancer Res. 58:2520-2523.