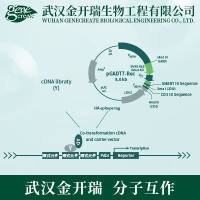
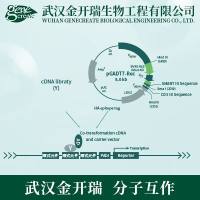
cDNA文库构建
互联网
1860
[ 阶段 3 : cDNA 的甲基化 ]
1. 在 cDNA 样品中加入以下试剂:
2 mol/L Tris-HCl (pH8.0) 5 μ l
5 mol/L NaCl 2 μ l
0.5 mol/L EDTA(pH8.0) 2 μ l
20 mmol/L S- 腺苷甲硫氨酸 1 μ l
加 H2 O 至 96 μ l
2. 取出两小份样品(各 2 μ l )至 0.5ml 微量离心管中,分别编为 1 号和 2 号,置于冰上。
3. 在余下的反应混合液中加入 2 μ l Eco R I 甲基化酶( 80 000 单位 /ml ),保存在 0 ℃直至步骤 4 完成。
4. 再从大体积的反应液中吸出另外两小分样品(各 2 μ l )至 0.5ml 微量离心管中,分别编为 3 号和 4 号。
5. 在所有四小份样品(来自步骤 2 和步骤 4 )加入 100 ng 质粒 DNA 或 500 ng 的λ噬菌体 DNA 。这些未甲基化的 DNA 在预实验中用作底物以测定甲基化效率。
6. 所有四份小样实验反应和大体积的反应均在 37 ℃温育 1h 。
7. 于 68 ℃加热 15min ,用酚:氯仿抽提大体积反应液一次,再用氯仿抽提一次。
8. 在大体积反应液中加入 0.1 倍体积的 3 mol/L 乙酸钠( pH5.2 )和 2 倍体积的乙醇,混匀后贮存于 -20 ℃直至获得小样反应结果。
9. 按下述方法分析 4 个小样对照反应:
a. 在每一对照反应中分别加入:
0.1 mol/L MgCl2 2 μ l
10 × Eco R I 缓冲液 2 μ l
加 H2 O 至 20 μ l
b. 在 2 号和 4 号反应管中分别加入 20 单位 Eco R I 。
c. 四个对照样品于 37 ℃温育 1h ,通过 1% 琼脂糖凝胶电泳进行分析。
10. 微量离心机以最大速度离心 15min(4 ℃ ) 以回收沉淀 cDNA 。弃上清,加入 200 μ l 70% 乙醇洗涤沉淀,重复离心。
11. 用手提式微型探测器检查是否所有放射性物质均被沉淀。小心吸出乙醇,在空气中晾干沉淀,然后将 DNA 溶于 29 μ l TE ( pH8.0 )。
12. 尽可能快地进行 cDNA 合成的下一阶段。
[ 阶段 4 :接头或衔接子的连接 ]
cDNA 末端的削平
1. cDNA 样品于 68 ℃加热 5 min 。
2. 将 cDNA 溶液冷却至 37 ℃并加入下列试剂:
5 × T4 噬菌体 DNA 聚合酶修复缓冲液 10 μ l
dNTP 溶液,每种 5 mmol/L 5 μ l
加 H2 O 至 50 μ l
3. 加入 1~2 单位 T4 噬菌体 DNA 聚合酶( 500 单位 /ml ), 37 ℃温育 15min 。
4. 加入 1 μ l 0.5mol/L EDTA(pH8.0) ,以终止反应。
5. 用酚:氯仿抽提,再通过 Sephadex G-50 离心柱层析,除去未掺入的 dNTP 。
6. 在柱流出液中加入 0.1 倍体积的 3 mol/L 乙酸钠( pH5.2 )和二倍体积的乙醇,样品于 4 ℃至少放置 15 min 。
7. 在微量离心机上以最大速度离心 15 min(4 ℃ ) ,回收沉淀的 cDNA 。沉淀经空气干燥后溶于 13 μ l 的 10 mmol/L Tris-HCl (pH8.0).
接头 - 衔接子与 cDNA 的连接
8. 将下列试剂加入到已削成平末端的 DNA 中:
10 × T4 噬菌体 DNA 聚合酶修复缓冲液 2 μ l
800~1000 ng 的磷酸化接头或衔接子 2 μ l
T4 噬菌体 DNA 连接酶( 105 Weiss 单位 /ml ) 1 μ l
10 mmol/L ATP 2 μ l
混匀后,在 16 ℃温育 8~12h 。
9. 从反应液中吸出 0.5 μ l 贮存于 4 ℃,其余反应液于 68 ℃加热 15min 以灭活连接酶。
[ 阶段 5 : Sepharose CL-4B 凝胶过滤法分离 cDNA]
Sepharose CL-4B 柱的制备
1. 用带有弯头的皮下注射针头将棉拭的一半推进 1ml 灭菌吸管端部,用无菌剪刀剪去露在吸管外的棉花并弃去,再用滤过的压缩空气将余下的棉拭子吹至吸管狭窄端。
2. 将一段无菌的聚氯乙烯软管与吸管窄端相连,将吸管宽端浸于含有 0.1 mol/L NaCl 的 TE ( pH7.6 )溶液中。将聚氯乙烯管与相连于真空装置的锥瓶相接。轻缓抽吸,直至吸管内充满缓冲液,用止血钳关闭软管。
3. 在吸管宽端接一段乙烯泡沫管,让糊状物静置数分钟,放开止血钳,当缓冲液从吸管滴落时,层析柱亦随之形成。如有必要,可加入更多的 Sepharose CL-4B ,直至填充基质几乎充满吸管为止。
4. 将几倍柱床体积的含 0.1 mol/L 氯化钠的 TE(pH7.6) 洗涤柱子。洗柱完成后,关闭柱子底部的软管。
依据大小分离回收 DNA
5. 用巴斯德吸管吸去柱中 Sepharose CL-4B 上层的液体,将 cDNA 加到柱上(体积 50 μ l 或更小),放开止血钳,使 cDNA 进入凝胶。用 50 μ l TE(pH7.6) 洗涤盛装 cDNA 的微量离心管,将洗液亦加于柱上。用含 0.1 mol/L NaCl 的 TE(pH7.6) 充满泡沫管。
6. 用手提式小型探测器监测 cDNA 流经柱子的进程。放射性 cDNA 流到柱长 2/3 时,开始用微量离心管收集,每管 2 滴,直至将所有放射性洗脱出柱为止。
7. 用切仑科夫计数器测量每管的放射性活性。
8. 从每一管中取出一小份,以末端标记的已知大小( 0.2kb~5kb )的 DNA 片断作标准参照物,通过 1% 琼脂凝胶电泳进行分析,将各管余下部分贮存于 -20 ℃,直至获得琼脂糖凝胶电泳的放射自显影片。
9. 电泳后将凝胶移至一张 Whatman 3MM 滤纸上,盖上一张 Saran 包装膜,并在凝胶干燥器上干燥。干燥过程前 20min 至 30min 于 50 ℃加热凝胶,然后停止加热,在真空状态继续干燥 1~2h.
10. 置 -70 ℃加增感屏对干燥的凝胶继续 X 射线曝光。
11. 在 cDNA 长度≥ 500bp 的收集管中,加入 0.1 倍体积的 3mol/L 乙酸钠( pH5.2 )和二倍体积的乙醇。于 4 ℃放置至少 15min 使 cDNA 沉淀,用微量离心机于 4 ℃以 12 000g 离心 15min ,以回收沉淀的 cDNA 。
12. 将 DNA 溶于总体积为 20 μ l 的 10 mmol/L Tris-HCl(pH7.6) 中。
13. 测定每一小份放射性活度。算出选定的组分中所得到的总放射性活度值。计算可用于λ噬菌体臂相连接的 DNA 总量。
[ 选定组分的总活度值 (cpm)/ 掺入到第二链的活度值 (cpm)~undefined2x μ g cDNA 第二链合成量 = 可用于连接的 cDNA
[NextPage]
[ 阶段 6 : cDNA 与λ噬菌体臂的连接 ]
1. 按下述方法建立 4 组连接 - 包装反应:
连接
A( μ l)
B( μ l)
C( μ l)
D( μ l)
λ噬菌体 DNA(0.5 μ g/ μ l)
1.0
1.0
1.0
1.0
10 × T4DNA 连接酶缓冲液
1.0
1.0
1.0
1.0
cDNA
0 ng
5 ng
10 ng
50 ng
T4 噬菌体 DNA 连接酶( 105 Weiss 单位 /ml )
0.1
0.1
0.1
0.1
10 mmol/L ATP
1.0
1.0
1.0
1.0
加 H2 O 至
10
10
10
10
连接混合物于 16 ℃培育 4~16h 。剩余的 cDNA 储存于 -20 ℃。
2. 按包装提取物厂商提供的方法,从每组连接反应物中取 5 μ l 包装到噬菌体颗粒中。
3. 包装反应完成后,在各反应混合物中加入 0.5ml SM 培养基。
4. 预备适当的大肠杆菌株新鲜过夜培养物,包装混合物做 100 倍稀释,各取 10 μ l 和 100 μ l 涂板,于 37 ℃或 42 ℃培养 8~12 小时。
5. 计算重组噬菌斑和非重组噬菌斑,连接反应 A 不应产生重组噬菌斑,而连接反应 B 、 C 和 D 应产生数目递增的重组噬菌斑。
6. 根据重组噬菌斑的数目,计算 cDNA 的克隆效率。
7. 挑取 12 个重组λ噬菌体空斑,小规模培养裂解物并制备 DNA ,以供适当的限制性内切核酸酶消化。
8. 通过 1% 琼脂凝胶电泳分析 cDNA 插入物的大小,用长度范围 500bp~5kb 的 DNA 片段作为分子质量参照。
[ 阶段 3 : cDNA 的甲基化 ] 1. 在 cDNA 样品中加入以下试剂: 2 mol/L Tris-HCl (pH8.0) 5 μ l 5 mol/L NaCl 2 μ l 0.5 mol/L EDTA(pH8.0) 2 μ l 20 mmol/L S- 腺苷甲硫氨酸 1 μ l 加 H2 O 至 96 μ l 2. 取出两小份样品(各 2 μ l )至 0.5ml 微量离心管中,分别编为 1 号和 2 号,置于冰上。 3. 在余下的反应混合液中加入 2 μ l Eco R I 甲基化酶( 80 000 单位 /ml ),保存在 0 ℃直至步骤 4 完成。 4. 再从大体积的反应液中吸出另外两小分样品(各 2 μ l )至 0.5ml 微量离心管中,分别编为 3 号和 4 号。 5. 在所有四小份样品(来自步骤 2 和步骤 4 )加入 100 ng 质粒 DNA 或 500 ng 的λ噬菌体 DNA 。这些未甲基化的 DNA 在预实验中用作底物以测定甲基化效率。 6. 所有四份小样实验反应和大体积的反应均在 37 ℃温育 1h 。 7. 于 68 ℃加热 15min ,用酚:氯仿抽提大体积反应液一次,再用氯仿抽提一次。 8. 在大体积反应液中加入 0.1 倍体积的 3 mol/L 乙酸钠( pH5.2 )和 2 倍体积的乙醇,混匀后贮存于 -20 ℃直至获得小样反应结果。 9. 按下述方法分析 4 个小样对照反应: a. 在每一对照反应中分别加入: 0.1 mol/L MgCl2 2 μ l 10 × Eco R I 缓冲液 2 μ l 加 H2 O 至 20 μ l b. 在 2 号和 4 号反应管中分别加入 20 单位 Eco R I 。 c. 四个对照样品于 37 ℃温育 1h ,通过 1% 琼脂糖凝胶电泳进行分析。 10. 微量离心机以最大速度离心 15min(4 ℃ ) 以回收沉淀 cDNA 。弃上清,加入 200 μ l 70% 乙醇洗涤沉淀,重复离心。 11. 用手提式微型探测器检查是否所有放射性物质均被沉淀。小心吸出乙醇,在空气中晾干沉淀,然后将 DNA 溶于 29 μ l TE ( pH8.0 )。 12. 尽可能快地进行 cDNA 合成的下一阶段。 [ 阶段 4 :接头或衔接子的连接 ] cDNA 末端的削平 1. cDNA 样品于 68 ℃加热 5 min 。 2. 将 cDNA 溶液冷却至 37 ℃并加入下列试剂: 5 × T4 噬菌体 DNA 聚合酶修复缓冲液 10 μ l dNTP 溶液,每种 5 mmol/L 5 μ l 加 H2 O 至 50 μ l 3. 加入 1~2 单位 T4 噬菌体 DNA 聚合酶( 500 单位 /ml ), 37 ℃温育 15min 。 4. 加入 1 μ l 0.5mol/L EDTA(pH8.0) ,以终止反应。 5. 用酚:氯仿抽提,再通过 Sephadex G-50 离心柱层析,除去未掺入的 dNTP 。 6. 在柱流出液中加入 0.1 倍体积的 3 mol/L 乙酸钠( pH5.2 )和二倍体积的乙醇,样品于 4 ℃至少放置 15 min 。 7. 在微量离心机上以最大速度离心 15 min(4 ℃ ) ,回收沉淀的 cDNA 。沉淀经空气干燥后溶于 13 μ l 的 10 mmol/L Tris-HCl (pH8.0). 接头 - 衔接子与 cDNA 的连接 8. 将下列试剂加入到已削成平末端的 DNA 中: 10 × T4 噬菌体 DNA 聚合酶修复缓冲液 2 μ l 800~1000 ng 的磷酸化接头或衔接子 2 μ l T4 噬菌体 DNA 连接酶( 105 Weiss 单位 /ml ) 1 μ l 10 mmol/L ATP 2 μ l 混匀后,在 16 ℃温育 8~12h 。 9. 从反应液中吸出 0.5 μ l 贮存于 4 ℃,其余反应液于 68 ℃加热 15min 以灭活连接酶。 [ 阶段 5 : Sepharose CL-4B 凝胶过滤法分离 cDNA] Sepharose CL-4B 柱的制备 1. 用带有弯头的皮下注射针头将棉拭的一半推进 1ml 灭菌吸管端部,用无菌剪刀剪去露在吸管外的棉花并弃去,再用滤过的压缩空气将余下的棉拭子吹至吸管狭窄端。 2. 将一段无菌的聚氯乙烯软管与吸管窄端相连,将吸管宽端浸于含有 0.1 mol/L NaCl 的 TE ( pH7.6 )溶液中。将聚氯乙烯管与相连于真空装置的锥瓶相接。轻缓抽吸,直至吸管内充满缓冲液,用止血钳关闭软管。 3. 在吸管宽端接一段乙烯泡沫管,让糊状物静置数分钟,放开止血钳,当缓冲液从吸管滴落时,层析柱亦随之形成。如有必要,可加入更多的 Sepharose CL-4B ,直至填充基质几乎充满吸管为止。 4. 将几倍柱床体积的含 0.1 mol/L 氯化钠的 TE(pH7.6) 洗涤柱子。洗柱完成后,关闭柱子底部的软管。 依据大小分离回收 DNA 5. 用巴斯德吸管吸去柱中 Sepharose CL-4B 上层的液体,将 cDNA 加到柱上(体积 50 μ l 或更小),放开止血钳,使 cDNA 进入凝胶。用 50 μ l TE(pH7.6) 洗涤盛装 cDNA 的微量离心管,将洗液亦加于柱上。用含 0.1 mol/L NaCl 的 TE(pH7.6) 充满泡沫管。 6. 用手提式小型探测器监测 cDNA 流经柱子的进程。放射性 cDNA 流到柱长 2/3 时,开始用微量离心管收集,每管 2 滴,直至将所有放射性洗脱出柱为止。 7. 用切仑科夫计数器测量每管的放射性活性。 8. 从每一管中取出一小份,以末端标记的已知大小( 0.2kb~5kb )的 DNA 片断作标准参照物,通过 1% 琼脂凝胶电泳进行分析,将各管余下部分贮存于 -20 ℃,直至获得琼脂糖凝胶电泳的放射自显影片。 9. 电泳后将凝胶移至一张 Whatman 3MM 滤纸上,盖上一张 Saran 包装膜,并在凝胶干燥器上干燥。干燥过程前 20min 至 30min 于 50 ℃加热凝胶,然后停止加热,在真空状态继续干燥 1~2h. 10. 置 -70 ℃加增感屏对干燥的凝胶继续 X 射线曝光。 11. 在 cDNA 长度≥ 500bp 的收集管中,加入 0.1 倍体积的 3mol/L 乙酸钠( pH5.2 )和二倍体积的乙醇。于 4 ℃放置至少 15min 使 cDNA 沉淀,用微量离心机于 4 ℃以 12 000g 离心 15min ,以回收沉淀的 cDNA 。 12. 将 DNA 溶于总体积为 20 μ l 的 10 mmol/L Tris-HCl(pH7.6) 中。 13. 测定每一小份放射性活度。算出选定的组分中所得到的总放射性活度值。计算可用于λ噬菌体臂相连接的 DNA 总量。 [ 选定组分的总活度值 (cpm)/ 掺入到第二链的活度值 (cpm)~undefined2x μ g cDNA 第二链合成量 = 可用于连接的 cDNA [NextPage] [ 阶段 6 : cDNA 与λ噬菌体臂的连接 ] 1. 按下述方法建立 4 组连接 - 包装反应: 连接 A( μ l) B( μ l) C( μ l) D( μ l) λ噬菌体 DNA(0.5 μ g/ μ l) 1.0 1.0 1.0 1.0 10 × T4DNA 连接酶缓冲液 1.0 1.0 1.0 1.0 cDNA 0 ng 5 ng 10 ng 50 ng T4 噬菌体 DNA 连接酶( 105 Weiss 单位 /ml ) 0.1 0.1 0.1 0.1 10 mmol/L ATP 1.0 1.0 1.0 1.0 加 H2 O 至 10 10 10 10 连接混合物于 16 ℃培育 4~16h 。剩余的 cDNA 储存于 -20 ℃。 2. 按包装提取物厂商提供的方法,从每组连接反应物中取 5 μ l 包装到噬菌体颗粒中。 3. 包装反应完成后,在各反应混合物中加入 0.5ml SM 培养基。 4. 预备适当的大肠杆菌株新鲜过夜培养物,包装混合物做 100 倍稀释,各取 10 μ l 和 100 μ l 涂板,于 37 ℃或 42 ℃培养 8~12 小时。 5. 计算重组噬菌斑和非重组噬菌斑,连接反应 A 不应产生重组噬菌斑,而连接反应 B 、 C 和 D 应产生数目递增的重组噬菌斑。 6. 根据重组噬菌斑的数目,计算 cDNA 的克隆效率。 7. 挑取 12 个重组λ噬菌体空斑,小规模培养裂解物并制备 DNA ,以供适当的限制性内切核酸酶消化。 8. 通过 1% 琼脂凝胶电泳分析 cDNA 插入物的大小,用长度范围 500bp~5kb 的 DNA 片段作为分子质量参照。
[ 阶段 3 : cDNA 的甲基化 ] 1. 在 cDNA 样品中加入以下试剂: 2 mol/L Tris-HCl (pH8.0) 5 μ l 5 mol/L NaCl 2 μ l 0.5 mol/L EDTA(pH8.0) 2 μ l 20 mmol/L S- 腺苷甲硫氨酸 1 μ l 加 H2 O 至 96 μ l 2. 取出两小份样品(各 2 μ l )至 0.5ml 微量离心管中,分别编为 1 号和 2 号,置于冰上。 3. 在余下的反应混合液中加入 2 μ l Eco R I 甲基化酶( 80 000 单位 /ml ),保存在 0 ℃直至步骤 4 完成。 4. 再从大体积的反应液中吸出另外两小分样品(各 2 μ l )至 0.5ml 微量离心管中,分别编为 3 号和 4 号。 5. 在所有四小份样品(来自步骤 2 和步骤 4 )加入 100 ng 质粒 DNA 或 500 ng 的λ噬菌体 DNA 。这些未甲基化的 DNA 在预实验中用作底物以测定甲基化效率。 6. 所有四份小样实验反应和大体积的反应均在 37 ℃温育 1h 。 7. 于 68 ℃加热 15min ,用酚:氯仿抽提大体积反应液一次,再用氯仿抽提一次。 8. 在大体积反应液中加入 0.1 倍体积的 3 mol/L 乙酸钠( pH5.2 )和 2 倍体积的乙醇,混匀后贮存于 -20 ℃直至获得小样反应结果。 9. 按下述方法分析 4 个小样对照反应: a. 在每一对照反应中分别加入: 0.1 mol/L MgCl2 2 μ l 10 × Eco R I 缓冲液 2 μ l 加 H2 O 至 20 μ l b. 在 2 号和 4 号反应管中分别加入 20 单位 Eco R I 。 c. 四个对照样品于 37 ℃温育 1h ,通过 1% 琼脂糖凝胶电泳进行分析。 10. 微量离心机以最大速度离心 15min(4 ℃ ) 以回收沉淀 cDNA 。弃上清,加入 200 μ l 70% 乙醇洗涤沉淀,重复离心。 11. 用手提式微型探测器检查是否所有放射性物质均被沉淀。小心吸出乙醇,在空气中晾干沉淀,然后将 DNA 溶于 29 μ l TE ( pH8.0 )。 12. 尽可能快地进行 cDNA 合成的下一阶段。 [ 阶段 4 :接头或衔接子的连接 ] cDNA 末端的削平 1. cDNA 样品于 68 ℃加热 5 min 。 2. 将 cDNA 溶液冷却至 37 ℃并加入下列试剂: 5 × T4 噬菌体 DNA 聚合酶修复缓冲液 10 μ l dNTP 溶液,每种 5 mmol/L 5 μ l 加 H2 O 至 50 μ l 3. 加入 1~2 单位 T4 噬菌体 DNA 聚合酶( 500 单位 /ml ), 37 ℃温育 15min 。 4. 加入 1 μ l 0.5mol/L EDTA(pH8.0) ,以终止反应。 5. 用酚:氯仿抽提,再通过 Sephadex G-50 离心柱层析,除去未掺入的 dNTP 。 6. 在柱流出液中加入 0.1 倍体积的 3 mol/L 乙酸钠( pH5.2 )和二倍体积的乙醇,样品于 4 ℃至少放置 15 min 。 7. 在微量离心机上以最大速度离心 15 min(4 ℃ ) ,回收沉淀的 cDNA 。沉淀经空气干燥后溶于 13 μ l 的 10 mmol/L Tris-HCl (pH8.0). 接头 - 衔接子与 cDNA 的连接 8. 将下列试剂加入到已削成平末端的 DNA 中: 10 × T4 噬菌体 DNA 聚合酶修复缓冲液 2 μ l 800~1000 ng 的磷酸化接头或衔接子 2 μ l T4 噬菌体 DNA 连接酶( 105 Weiss 单位 /ml ) 1 μ l 10 mmol/L ATP 2 μ l 混匀后,在 16 ℃温育 8~12h 。 9. 从反应液中吸出 0.5 μ l 贮存于 4 ℃,其余反应液于 68 ℃加热 15min 以灭活连接酶。 [ 阶段 5 : Sepharose CL-4B 凝胶过滤法分离 cDNA] Sepharose CL-4B 柱的制备 1. 用带有弯头的皮下注射针头将棉拭的一半推进 1ml 灭菌吸管端部,用无菌剪刀剪去露在吸管外的棉花并弃去,再用滤过的压缩空气将余下的棉拭子吹至吸管狭窄端。 2. 将一段无菌的聚氯乙烯软管与吸管窄端相连,将吸管宽端浸于含有 0.1 mol/L NaCl 的 TE ( pH7.6 )溶液中。将聚氯乙烯管与相连于真空装置的锥瓶相接。轻缓抽吸,直至吸管内充满缓冲液,用止血钳关闭软管。 3. 在吸管宽端接一段乙烯泡沫管,让糊状物静置数分钟,放开止血钳,当缓冲液从吸管滴落时,层析柱亦随之形成。如有必要,可加入更多的 Sepharose CL-4B ,直至填充基质几乎充满吸管为止。 4. 将几倍柱床体积的含 0.1 mol/L 氯化钠的 TE(pH7.6) 洗涤柱子。洗柱完成后,关闭柱子底部的软管。 依据大小分离回收 DNA 5. 用巴斯德吸管吸去柱中 Sepharose CL-4B 上层的液体,将 cDNA 加到柱上(体积 50 μ l 或更小),放开止血钳,使 cDNA 进入凝胶。用 50 μ l TE(pH7.6) 洗涤盛装 cDNA 的微量离心管,将洗液亦加于柱上。用含 0.1 mol/L NaCl 的 TE(pH7.6) 充满泡沫管。 6. 用手提式小型探测器监测 cDNA 流经柱子的进程。放射性 cDNA 流到柱长 2/3 时,开始用微量离心管收集,每管 2 滴,直至将所有放射性洗脱出柱为止。 7. 用切仑科夫计数器测量每管的放射性活性。 8. 从每一管中取出一小份,以末端标记的已知大小( 0.2kb~5kb )的 DNA 片断作标准参照物,通过 1% 琼脂凝胶电泳进行分析,将各管余下部分贮存于 -20 ℃,直至获得琼脂糖凝胶电泳的放射自显影片。 9. 电泳后将凝胶移至一张 Whatman 3MM 滤纸上,盖上一张 Saran 包装膜,并在凝胶干燥器上干燥。干燥过程前 20min 至 30min 于 50 ℃加热凝胶,然后停止加热,在真空状态继续干燥 1~2h. 10. 置 -70 ℃加增感屏对干燥的凝胶继续 X 射线曝光。 11. 在 cDNA 长度≥ 500bp 的收集管中,加入 0.1 倍体积的 3mol/L 乙酸钠( pH5.2 )和二倍体积的乙醇。于 4 ℃放置至少 15min 使 cDNA 沉淀,用微量离心机于 4 ℃以 12 000g 离心 15min ,以回收沉淀的 cDNA 。 12. 将 DNA 溶于总体积为 20 μ l 的 10 mmol/L Tris-HCl(pH7.6) 中。 13. 测定每一小份放射性活度。算出选定的组分中所得到的总放射性活度值。计算可用于λ噬菌体臂相连接的 DNA 总量。 [ 选定组分的总活度值 (cpm)/ 掺入到第二链的活度值 (cpm)~undefined2x μ g cDNA 第二链合成量 = 可用于连接的 cDNA [NextPage] [ 阶段 6 : cDNA 与λ噬菌体臂的连接 ] 1. 按下述方法建立 4 组连接 - 包装反应: 连接 A( μ l) B( μ l) C( μ l) D( μ l) λ噬菌体 DNA(0.5 μ g/ μ l) 1.0 1.0 1.0 1.0 10 × T4DNA 连接酶缓冲液 1.0 1.0 1.0 1.0 cDNA 0 ng 5 ng 10 ng 50 ng T4 噬菌体 DNA 连接酶( 105 Weiss 单位 /ml ) 0.1 0.1 0.1 0.1 10 mmol/L ATP 1.0 1.0 1.0 1.0 加 H2 O 至 10 10 10 10 连接混合物于 16 ℃培育 4~16h 。剩余的 cDNA 储存于 -20 ℃。 2. 按包装提取物厂商提供的方法,从每组连接反应物中取 5 μ l 包装到噬菌体颗粒中。 3. 包装反应完成后,在各反应混合物中加入 0.5ml SM 培养基。 4. 预备适当的大肠杆菌株新鲜过夜培养物,包装混合物做 100 倍稀释,各取 10 μ l 和 100 μ l 涂板,于 37 ℃或 42 ℃培养 8~12 小时。 5. 计算重组噬菌斑和非重组噬菌斑,连接反应 A 不应产生重组噬菌斑,而连接反应 B 、 C 和 D 应产生数目递增的重组噬菌斑。 6. 根据重组噬菌斑的数目,计算 cDNA 的克隆效率。 7. 挑取 12 个重组λ噬菌体空斑,小规模培养裂解物并制备 DNA ,以供适当的限制性内切核酸酶消化。 8. 通过 1% 琼脂凝胶电泳分析 cDNA 插入物的大小,用长度范围 500bp~5kb 的 DNA 片段作为分子质量参照。