在大肠杆菌中高效表达外源蛋白的全攻略
互联网
蛋白表达技术全攻略
本世纪60至70年代对大肠杆菌的研究使之成为自然界中最普遍为人们所认识的生物体。大肠杆菌具有两个显著特征:操作简单和能在廉价的培养基中高密度培养,它的这些特征加上十多年外源基因表达的经验使其在大多数科研应用中成为高效表达异源蛋白最常用的原核表达系统。尽管大肠杆菌有众多的优点,但并非每一种基因都能在其中有效表达。这归因于每种基因都有其独特的结构、mRNA的稳定性和翻译效率、蛋白质折叠的难易程度、宿主细胞蛋白酶对蛋白质的降解、外源基因和E.coli 在密码子利用上的主要差别以及蛋白质对宿主的潜在毒性等等。但知识的大量积累还是有助于为表达方面某些特定的困难提供一般的解决方法。
大肠杆菌作为表达系统的主要障碍包括:不能象真核蛋白那样进行翻译后修饰、缺乏将蛋白质有效释放到培养基中的分泌机制和充分形成二硫键的能力。
另一方面,许多真核蛋白在非糖基化的形式下能保留其生物学活性,因而也就可以用大肠杆菌来表达。如何实现外源基因在原核细胞中的有效表达,自60年代以来,对影响外源基因在其表达体系中表达效率的各个因素作了大量实验研究,并有多篇归纳性综述发表[1,2,3]。国内针对外源基因在原核细胞中高效表达的关键因素,构建了高效表达载体[4],并在此基础上成功表达了一系列细胞因子的基因[5,6,7]。我们在分析了国内外有关在原核系统中表达蛋白的实验资料的基础上,对在大肠杆菌中高效表达外源蛋白的策略所涉及的内容进行全面的总结,以期有助于我国在这方面的研究。
一.有效表达载体的构型
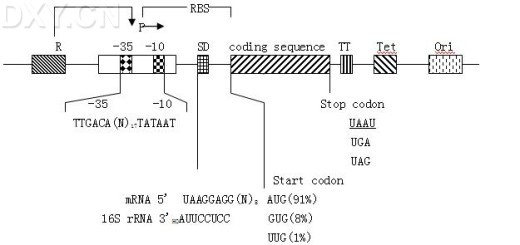
构建表达质粒需要多种元件,需要仔细考虑它们的组合,以保证最高水平的蛋白质合成 。E.coli 表达载体的基本结构[8]。
<center> </center>启动子(以杂和的tac 启动子为例)位于核糖体结合位点(RBS)上游10-100bp处,由调节基因(R)控制,调节基因可以是载体自身携带,也可以整合到宿主染色体 上。E.coli 的启动子由位于转录起始位点上游约35bp的六核苷酸序列(-35区)和一短序列隔开的另一六核苷酸序列(-10区)组成[9,10,11]。有许多启动子可用于在E.coli中的基因表达,包括来源于革兰氏阳性菌和噬菌体的启动子。理想的启动子具有以下特性:作用强;可以严格调控;容易转导入其他E.coli 以便筛选大量的用于生产蛋白的菌株,而且对其诱导是简便和廉价的[12]。在启动子下游是RBS,其跨度约为54个核苷酸,两端限定在 -35(±2)和mRNA编码序列的+19到+22之间[13]。
Shine-Dalgarno(SD)位点[14,15]在翻译起始阶段与16S rRNA的3’端相互作用[16]。SD与起始密码子间的距离约为5-13bp[17],而且此区的序列在mRNA转录物中应避免出现二级结构,否则将会降低翻译起始的效率[18]。在RBS的5’和3’端均为A丰富区[19]。转录终止子位于编码序列的下游,作为转录终止的信号[20]和组成发卡结构的保护性元件,阻止核酸外切酶对mRNA的降解,从而延长mRNA的半衰期[21]。
除了上述对基因表达的效率有直接影响的元件以外,载体还含有抗生素抗性基因,以方便质粒的筛选和传代。氨苄青霉素是最常用的抗性标记。但在生产人用治疗性蛋白时,最好选择其他抗性标记(如Tet)以避免可能发生的过敏反应[22]。质粒的拷贝数由复制起点决定。在特殊情况下,选用失控复制子能够获得大量的质粒拷贝数和较高产量的质粒编码蛋白[23,24]。但在另外一些情况下,选用超过pBR322的高拷贝质粒似乎并没有好处。而且已有资料表明,增加质粒的拷贝数降低E.coli中胰酶的产量,在高拷贝质粒中,强启动子的存在严重影响了细胞的活性[25,26]。
二.转录水平调控
1.启动子
能在E.coli中发挥作用的启动子很多。这些启动子必须具有适合高水平蛋白质合成的某些特性。首先启动子的作用要强,待表达基因的产物要占或超过菌体总蛋白的的10-30%;第二,它必须表现最低水平的基础转录活性。若要求大量的基因表达,最好选用高密度培养细胞和表现最低活性的可诱导和非抑制启动子。如果所表达的蛋白具有毒性或限制宿主细胞的生长,选用可抑制的启动子则至关重要[27,28]。
例如,轮状病毒的VP7蛋白能有效地杀死细胞,因此必须在严格控制的条件下表达[29]。但在某些情况下,启动子的严格性并不合理,因为即使最小量的基因产物也能由于其毒性而杀死细胞。如能灭活核糖体或破坏膜渗透压的分子对细胞来说是致死性的[30]。对宿主的毒性并不仅限于外源基因,某种自身蛋白的过量表达也能造成同样的结果。如编码外膜磷脂蛋白的traT基因[31]。
另外,不完全抑制的表达系统会造成质粒的不稳定,细胞生长速度的下降和重组蛋白产量的降低[32,33]。Lanzer和Bujard曾对常用的lac启动子-操纵子系统进行了广泛的研究,证明操纵子放在启动子序列的不同位置会造成70倍差异的抑制。将17bp的操纵子置于-10和-35六聚体区之间所形成的抑制比将其放在-35区的上游或-10区的下游要高50-70倍[34]。启动子的第三个特性是其简便和廉价的可诱导性。
大量生产蛋白质最常用的启动子是热诱导(λPL)和化学诱导(trp)启动子。异丙基硫代-β-D-半乳糖苷(IPTG)诱导的杂交启动子tac[35]或trc[36]都是强启动子,在基础研究中应用很广。但在大量生产人用治疗性蛋白时用IPTG做诱导剂是不可取的,因为IPTG具有毒性而且价格昂贵[37]。IPTG的这些不足至今仍限制tac或trc强启动子在大量生产人用治疗性蛋白中的应用。编码热敏lac阻遏蛋白[38]的突变lacI(Ts)基因的出现使得目前能够对这些启动子进行热诱导[39,40]。另外,还出现了一些新型载体,它们允许在30℃对trc启动子进行严紧调节。
最近还报道了两种不同的lac阻遏蛋白突变体,能够同时允许热诱导和IPTG诱导[41,42]。尽管野生型lacI基因也能热诱导,但不能对其进行严紧型调节,且不能用于lacIq 菌株,因为温度的变化不能遏制由lac阻遏蛋白的过量表达所造成的严紧性抑制[43]。因此,该系统只能用用于生产对宿主菌无害的一些蛋白。
冷反应启动子尽管不象其他启动子那样得到广泛的研究,但已经被证明能在低温条件下进行有效的基因表达。噬菌体λPL启动子的活性在20℃时最高,随着温度的升高,其活性逐渐降低[44]。PL启动子的冷反应由E.coli整合宿主因子,一种与DNA结合的序列特异性多功能蛋白来正调控[45,46]。主要冷应激基因cspA启动子同样也被证明在低温具有活性[47]。
对cspA和PL启动子进行分子剖析,鉴定了在较低温度下参与增强转录的特异性DNA区域。从而开发了一系列在20℃低温具有高活性的PL衍生启动子[48]。选用冷反应启动子的基本原理是在15-20℃的条件下,蛋白质的折叠速度只受到轻微的影响,而作为生物化学反应,转录和翻译的速度将被充分降低。这就为蛋白质折叠、产生有活性的蛋白和避免非活性蛋白聚合体即包涵体的形成提供了充足的时间,而目的蛋白的最终产量并未减少。
最近新报道的一些启动子具有诸多诱人之处,为选择新的高效表达系统提供了方便。如非常强的pH启动子[49,50],重组蛋白的产量可达总蛋白的40-50%[51]。但表达的水平会因不同的基因而有差异。因为蛋白质的合成不仅依赖于启动子的强弱,而且有赖于转录的效率。
人们通常只考虑E.coli启动子的核心区域,即-10和-35六核苷酸区和一15-19bp的间隔序列。但有人提出核心序列以外的元件也能刺激启动子的活性[52]。许多研究也已证明,核心启动子上游的序列能够在体内提高转录起始的效率[53,54,55]。Gourse等证明,位于E.coli rRNA启动子rrnB P1 -35区上游的DNA序列即UP元件,能够在体内和体外提高转录效率30倍[56]。
UP元件是作为一个独立的启动子组件发挥作用的,因为将其融合到其他启动子如lacUV5中也能刺激转录[57,58]。UP元件与其他启动子融合所表现的这种转录增强子效应,使其有望通用于高效表达系统。尽管已经证明rRNA启动子P1、P2的超强能力[59],但它们仍未被用于E.coli进行高水平表达,其主要原因是难以对其进行调控。rRNA的体内合成从属于细胞增殖速度的控制。
在细胞快速增殖期,P1和P2是活化的,当细胞处于生长的静息期,则P1、P2被负调节。因此,rRNA启动子在前诱导期将持续活化。在体内快速增殖的细胞中,P2的活性很弱,而且可诱导性差,但若与P1分开,P2启动子的活性明显升高(达到P1的70%),且对应激反应敏感。这表明在天然的串联体中,P2是部分关闭的[60]。Brosius和Holy[61]将lac操纵子序列插入到rrnB rRNA P2启动子的下游,在带有lacIq基因的菌株中成功地抑制了P2的活性。
转录活性是以氯霉素乙酰转移酶的产生和4.5s RNA的表达为标准的。然而,P2构建体的活性只相当于tac启动子活性的1/2,而且当rrnB P1启动子被置于P2启动子下游时,不能完全抑制转录。有人试图利用反转启动子的方向来严格调控rRNA启动子。将rRNA启动子克隆于目的基因的上游,但转录方向相反。利用λ整合位点和可调控的λ整合酶来实现启动子的反转,从而达到进行诱导的目的,而且,在高度活化的启动子上游有一强转录终止子将避免在前诱导期可能出现的载体的不稳定性。
2.转录终止子
在原核生物中,转录终止有两种不同的机制。一种是依赖六聚体蛋白rho的rho依赖性转录终止,rho蛋白能使新生RNA转录本从模板解离。另一种是rho非依赖性转录终止,它特异性依赖于模板上编码的信号,即在新生RNA中形成发卡结构的一回文序列区,和位于该回文序列下游4-9bp处的dA、dT富含区[62,63]。虽然转录终止子在表达质粒的构建过程中常被忽略,但有效的转录终止子是表达载体必不可少的元件,因为它们具有极其重要的作用。贯穿启动子的转录将抑制启动子的功能,造成所谓的启动子封堵[64]。
这种效应可以通过在编码序列下游的适当位置放置一转录终止子,阻止转录贯穿别的启动子来避免。同样地,在启动目的基因的启动子上游放置一转录终止子,将最大限度地减小背景转录[65]。另有资料证明,由强启动子启动的转录会使转录通读到复制区,造成控制质粒拷贝数的ROP蛋白的过量表达,从而导致质粒的不稳定[66]。另外,转录终止增强mRNA的稳定性[67],从而提高蛋白质的表达水平。如果来源于E.coli rrB rRNA操纵子的两个转录终止子T1和T2串联,则效果更好[68]。但其他的许多转录终止子通常情况下都是十分有效的。
3.转录抗终止子
细菌中许多参与氨基酸生物合成的操纵子在其第一个结构基因的5’端含有转录衰减子。衰减子由特定操纵子的氨基酸产物调节。这样关联的带电荷tRNA的存在就会在核糖体剪切之后引导初始转录本中二级结构的形成。如果没有关联的带电荷tRNA,则会形成抗终止子结构,从而抑制终止子中发卡结构的形成和转录的终止[69]。抗终止元件能使RNA 多聚酶越过核糖体RNA操纵子中的rho依赖性终止子即boxA[70,71]。转录抗终止是有一个极其复杂的过程,其中包含许多已知和未知因子。有关这方面的知识已有详尽的综述。在此我们主要讨论抗终止元件在E.coli中表达外源基因的应用。
在E.coli表达系统中作用比较强、应用较广的一个启动子是噬菌体T7晚期启动子[72,73]。该系统的活性依赖于提供T7 RNA多聚酶的转录单元。RNA多聚酶的严格阻遏对防止T7启动子的渗漏是必需的。有许多用于调节T7多聚酶表达的途径,但每一种方法都有其各自的缺陷。Merten等通过构建一基于λPL调节的反转衰减T7 RNA多聚酶表达盒阐明了这一问题。可以通过在启动子和编码T7多聚酶的基因之间插入三个串联排列的转录终止子来削弱T7多聚酶的基础表达水平。
在诱导的情况下,噬菌体λ来源的nutL依赖的抗终止功能也可以协同来阻止转录终止[74]。来源于E.colirrnB rRNA操纵子的转录抗终止区已被用于某些表达载体如pSE420。该载体应用trc启动子[75]。其基本原理是使得转录通过重度二级结构区,从而减少宿主RNA 多聚酶引起的转录提前终止的可能性。但是rrnB抗终止子在这种情况下似乎并不十分有效。
4.严紧型调节表达系统
可严紧调节的启动子的优点使得我们能够设计许多巧妙、可高度抑制的表达系统。这在表达那些产物对宿主的生长不利的基因时尤其有用。所用的策略包括“铺平板”法[76];提高抑制基因和操纵基因的比例[77];用突变的噬菌体感染诱导[28 ];致弱高拷贝数载体上的启动子活性[78];利用转录终止子和抗终止子[79];在拷贝数可控的质粒中使用可诱导的启动子[80];使用“交叉调节”系统[81];共转导使用SP6 RNA多聚酶的质粒[82];利用与克隆基因mRNA互补的反义RNA[31]。最后一个巧妙的方法是反转启动子:在启动子两侧为两个λ整合位点,启动子的方向与基因的表达方向相反,而且只通过由λ整合酶介导的诱导位点特意性遗传重组来完成反转[83,84]。
上述这些系统也各有其优缺点。即依赖固体培养基的方法不适用于大批量表达,高水平抑制系统常造成蛋白质产量的下降[85],这就有必要优化抑制基因和操纵基因的比例[86]。由λ噬菌体介导的诱导更增加了系统的复杂性,利用反转启动子迂回方法又引入了复杂的载体结构。尽管这些表达系统大多数尚未用于蛋白质的大规模、高产量生产,但是它们为基因表达提供了重要的工具。
三、翻译水平的调控
1.mRNA翻译起始
有关转录过程的大量知识使得能够在不被附近核苷酸序列影响的情况下,在表达盒中使用原核启动子[87]。对于决定蛋白质合成的起始因素,虽然尚不能完全弄清楚,但目前已经明白,mRNA转录本5’末端的独特结构是mRNA翻译起始效率的最主要决定因素。至今还没有发现通用的有效起始翻译的共同序列。已知序列的绝大部分(91%)E.coli基因的翻译起始区均含有起始密码子AUG,GUG的利用率为8%,而UUG的利用率则为1%[88,89]。Shine-Dalgarno(SD)位点在翻译起始阶段与16S rRNA的3’端相互作用。SD与起始密码子间的距离约为5-13bp,这一距离影响翻译起始的效率[90]。人们对此进行了深入的研究,以确定最佳的SD区序列和SD与起始密码子之间的最有效间隔[91,92]。
Ringquist等[93]研究了RBS的翻译功能,得出了以下结论:(1)在间隔相同的情况下,UAAGGAGG的SD序列比AAGGA的SD序列能使蛋白质的产量提高3-6倍;(2)对于同一SD序列,存在一最佳的间隔。AAGGA的间隔为5-7个核苷酸,而UAAGGAGG的间隔为4-8个核苷酸;(3)对于同一SD序列,有一翻译所必需的最小间隔。AAGGA的最小间隔为5个核苷酸,而UAAGGAGG的最小间隔为3-4个核苷酸。这些间隔提示,在16S rRNA的3’末端和结合于核糖体P位点的fMet-tRNAf的反义密码子之间存在精确的物理关系。
在mRNA的翻译起始区的二级结构在决定基因表达效率方面具有重要作用[94,95,96]。如用一发卡结构封闭SD区和/或AUG密码子就会阻止其与30S核糖体亚单位的结合,从而抑制翻译[97]。已经设计了几种不同的策略以使mRNA形成二级结构的可能性最小。提高RBS中A、T残基的丰度能增强某些基因的表达[98]。同样地,突变SD区上游或下游的某些特定核苷酸就会抑制mRNA二级结构的形成和提高翻译效率[99]。
另一途径得益于在E.coli中自然发生的一种翻译偶联现象[100]。翻译偶联的机制已被用来解释来自多顺反子mRNA的不同基因的并列表达。已经证明,当galK与上游基因偶联翻译时,经修饰的gal强启动子能够指导半乳糖激酶的高水平合成。这提示即使很弱的RBS,如果与核糖体结合也能非常有效。这种调节机制有可能在蛋白质超量生产生物技术中发挥重要作用。事实上,翻译偶联已经被广泛用于不同基因的高效表达。
除了SD区与16S rRNA结合之外,mRNA和核糖体的其他相互作用也参与翻译的起始。如核糖体S1蛋白直接参与30S亚基对mRNA的识别和结合[101]。
2.翻译增强子
已经在细菌和噬菌体中鉴定了一些在E.coli中显著增强异源基因表达的序列。Olins等从T7噬菌体基因10前导序列(g10-L)中鉴定了一9bp的序列,该序列似乎能替代有效的RBS。同SD共有序列相比,g10-L能使多种基因的表达水平提高40-340倍[102]。若将其置于合成SD序列的上游,按照β-半乳糖苷酶的活性与LacZ mRNA的水平来估计,g10-L序列能使 LacZ的翻译水平提高110倍[103]。
另外的研究小组在mRNA的5’非翻译区(UTR)鉴定了一U富含序列,该序列同样具有翻译增强子活性。McCarthy等[104]在 E.coli atpE基因中紧接于SD位点下游鉴定一类似区域。有文献用一30bp的序列超量产生IL-2和IFN-β[105]。另有人证明在编码RNase D的rnd mRNA SD位点的上游,有一U8序列对该mRNA的有效翻译是必需的。缺失这一区域会显著降低翻译,但不影响rnd mRNA的水平和转录起始位点[106]。研究证明这些类似序列的靶位是30S核糖体亚单位的S1蛋白[107]。
在另一项有趣的研究中,研究者证明在紧接起始密码子下游的序列在翻译起始过程中发挥重要作用。位于T7基因0.3编码区+15至+26之间或位于T7基因10编码区+9至+21之间被称做下游框(DB)的特定区域具有翻译增强子的功能。DB区与16S rRNA的1469-1483核苷酸互补,这一区域称做反下游框(ADB)。缺失DB将废除翻译活性。相反,如果优化DB和ADB之间的互补则会以最高水平表达dhfr融合基因。有趣的是,如果将DB从起始密码子的上游移到SD序列的位置,DB则失去功能。DB序列存在于一些E.coli和噬菌体基因中[108,109]。
上述这些发现充分证明,除了SD位点和起始密码子以外,mRNA中的其他序列对于有效的翻译也是重要的。尽管其精确的机制还不太清楚,但有可能利用翻译增强子来达到超量表达蛋白质的目的。
3.mRNA的稳定性
mRNA的快速降解势必影响蛋白质的产生。因此在这一部分重点阐述决定mRNA稳定性的因素,这将在E.coli高效表达外源基因中有实际应用。在E.coli中有多种不同的RNase参与mRNA的降解,其中包括内切核酸酶(RNase E,RNase K和RNase III)和3’外切核酸酶(RNase II和多聚核苷酸磷酸化酶[PNPase]),目前尚未在原核细胞中发现5’外切核酸酶[110]。mRNA的降解并非由非特异性的外切核酸酶随机剪切而引起,因为在mRNA的长度和半衰期之间并没有反向相关性[111]。
已经证明,在E.coli中有两类保护性元件能够稳定mRNA。一类由mRNA的5’UTRs中的序列组成[112];另一类由3’UTRs和多顺反子间区的发卡结构组成[113]。其中一些元件与异源mRNA融合后起稳定剂作用,但只在严格的条件下如此。例如,噬菌体T4基因32的5’UTR在T4噬菌体感染的细胞中延长非稳定mRNA在E.coli中的半衰期[114]。革兰氏阳性菌如金黄色葡萄球菌和枯草杆菌的红霉素抗性基因(erm)编码的mRNA 5’UTR含有稳定元件。但ermC和ermA 5’UTRs的稳定作用需要由抑制翻译和引起核糖体失控的抗生素来诱导[115]。同样,噬菌体λPL对于λPL-trp转录本的稳定作用需要λ噬菌体的感染[116]。
与此相反,E.coli ompA转录本能够在细胞快速增殖的正常情况下延长一系列异源mRNA在E.coli中的稳定性[117]。Emory等证明,在接近或紧接ompA 5’UTR的5’末端存在发卡结构对于其稳定效果是必需的。而且可以通过在5’末端添加发卡结构来延长在正常情况下不稳定的mRNA的半衰期[118]。这样看来,对异源基因添加ompA 5’稳定元有可能提高E.coli中的基因表达。
另一类由3’UTR组成的mRNA保护性元件能够形成发卡结构,因而能够阻断外切核酸酶从3’末端对转录本的降解[113]。Wong和Chang[119]在苏云金杆菌的晶体蛋白基因的转录终止子中鉴定了一个这样的元件。将该“阳性反调节子”与地衣杆菌的青霉素酶基因(penP)的3’末端和人IL-2 cDNA融合,能够延长mRNA的半衰期,且提高了相应多肽在枯草杆菌和E.coli中的产量。然而,同某些5’稳定元一样,这类3’反向调节子不可能作为一个通用的mRNA稳定元。而且有证据表明,可以通过选用缺乏某些特定RNase如RNaseII或PNPase的宿主菌来提高基因的表达。这同样并非一有效途径。因为缺乏RNaseII或PNPase与RNase过量表达一样,对于E.coli整体mRNA的平均半衰期并无多大影响。而且缺乏RNaseII或PNPase的菌株常常是不稳定的[120,121]。
4.翻译终止
在mRNA中存在终止信号是翻译终止过程必不可少的。除了三个终止密码子UAA、UGA、
和UAG外,翻译终止这一复杂事件还包括核糖体、mRNA和终止位点的几种释放因子的特异相互作用[122]。在E.coli中,RF-1在终止密码子UGA处终止翻译;RF-2在UAA密码子处终止翻译[123]。最近还克隆了另外一个因子RF-3[124]。
在设计表达载体时,通常插入三个终止密码子以防止核糖体的跳跃。在E.coli中偏向于使用UAA密码子[125]。一项对于2000多个E.coli基因的统计分析表明,在终止密码子和紧接三联体的核苷酸序列中存在局部非随机性[126]。同时他们还利用体内终止试验测定12个可能的四核苷酸“终止信号”(UAAN、UGAN、UAGN)的终止力量。
在体内终止试验中,通过其与框架移位的竞争测定其终止效率。转录效率依据终止密码子和第四个核苷酸而有显著的差异,这种差异从80%(UAAU)到7%(UGAC)不等。这些研究表明,紧接终止密码子后的核苷酸特性强烈地影响E.coli中的翻译终止效率[127]。UAAU是E.coli中最有效的转录终止序列。此外,终止密码子5’末端的邻近序列也影响终止的效率。因此,新生肽中倒数第二位(-2位)C-末端氨基酸残基的电荷和疏水性能引起多达30倍不同的UGA终止效率,而在UAG位的终止对于-2位氨基酸残基的特性不敏感[128]。对于-1位,α-螺旋、β-链和回转倾向是UGA终止中的决定因素[129]。
四、蛋白质靶向
确定将目的蛋白表达在特定的细胞室,即细胞质、细胞间质或是培养基中,需要权衡各自的利弊。
1.细胞质表达
包涵体的形成仍然是在细胞质中进行基因表达的一个主要障碍。包涵体虽然具有多方面的好处,但这些优越之处与后期繁琐的蛋白重新折叠工作、重叠蛋白的生物活性的未知性以及重叠和纯化后蛋白的总产量降低相比,则显得微不足道。至今尚不明了包涵体形成的精确机制。对于形成和不形成包涵体的81种蛋白质的统计学分析表明,有6个主要的理化指标与此有关。这6个指标是电荷平均数、转变形成的残基组分、半胱氨酸组分、脯氨酸组分、亲水性和残基总数。其中前两个指标与包涵体形成密切相关。一种基于这些指标的模型可以根据某种蛋白质的氨基酸组成来预测包涵体形成的可能性。该模型曾成功地预测人T细胞受体Vβ5.3在E.coli中的可溶性[130] 。
已经建立了多种策略以帮助蛋白质天然空间结构的形成[131]。这些策略包括在较低温度下培养细菌[132],选择不同的E.coli菌株[133],替换某些氨基酸残基[134],与分子伴侣共表达[135,136,137],利用高溶解性的多肽作为共表达分子[138],在山梨糖醇和甘氨酸三甲内盐存在时,以低渗透压培养和诱导细胞[139],改变培养基的pH值[140]。
与分子伴侣共表达或许是一种有望提高蛋白质可溶性和折叠效率的途径[141]。但这种策略似乎具有蛋白质特异性[142]。即便在分子伴侣存在的条件下,仍有多种因素使得过量表达的蛋白不能折叠成其天然构象。这些因素包括缺乏二硫键和/或翻译后修饰;细胞质的氧化还原状态妨碍了二硫键的形成。在E.coli中,有两条途径参与二硫键的还原。即硫氧还蛋白系统,该系统由硫氧还蛋白还原酶和硫氧还蛋白组成;谷氧还蛋白系统,该系统由谷胱甘肽还原酶、谷胱甘肽和三种谷氧还蛋白组成[143]。制造次还原细胞质环境,以利于二硫键形成的策略包括选用硫氧还蛋白还原酶(trxB)缺陷的E.coli菌株,这有助于巯基还原势能。
蛋白质在细胞质中被降解的可能性比其他细胞室要大得多。因为在细胞质中含有大量的蛋白酶。另外,从细胞质蛋白混合物中纯化目的蛋白相对比较困难,因为在此细胞室包含总细胞蛋白的绝大部分蛋白[144]。
2.细胞外周质表达
在细胞外周质进行蛋白质表达有许多优越之处。在外周质只有4%的总细胞蛋白,这显然有利于目的蛋白的纯化,外周质的氧化环境有利于蛋白质的正确折叠,在转移到外周质的过程中,信号肽在细胞内剪切更有可能产生目的蛋白的天然N-末端。此外,外周质中的蛋白质降解也少得多[145]。
蛋白质通过内膜转运到外周质需要信号肽[146,147,148]。许多原核和真核细胞来源的信号肽已成功地用于Ecoli中蛋白质从内膜到外周质的转运。如E.coli的PhoA信号[149]、OmpA[150]、OmpT[151]、LamB和OmpF[152]以及金黄色葡萄球菌的A蛋白[153],鼠RNase[154]和人生长激素信号肽[155]等。但是,蛋白质转运到细菌外周质是一个特别复杂和尚未完全明了的过程,信号肽的存在并不总能保证有效的蛋白质通过内膜转运[156]。改善蛋白质转运到外周质的策略包括提供蛋白质转运和加工所需的成分:过量表达信号肽酶I[157],利用prlF突变株[158],共表达参与膜转运的几种蛋白质,降低蛋白质的表达水平以防止转运工具的过载[159]。
3. 细胞外分泌(外泌)
将蛋白质分泌到细胞外是人们最期望的一种策略。因为这样容易纯化目的蛋白质,减少细菌的蛋白酶对目的蛋白质的裂解。但是,E.coli在正常情况下只有很少量的蛋白质分泌到细胞外。要解决蛋白质外泌方面的难题,必须弄清E.coli的分泌途径。Pugsley[160]对革兰氏阴性菌的分泌途径进行了详细的研究。
在E.coli中将蛋白质分泌到培养基中的方法大致分为两类:(1)利用已有的“真正”的分泌蛋白所采用的途径[161];(2)利用信号肽序列、融合伴侣和具有穿透能力的因子。
第一种方法具有将目的蛋白质特异性分泌的优点,并最小限度地减少了非目的蛋白的污染。最突出的例子是溶血素基因,该基因曾被用于构建分泌的杂交蛋白[162,163];第二种方法依赖于有限渗透的诱导而导致蛋白质的分泌。例如应用pelB[164]、ompA[165]和A蛋白引导序列[153,166];与细菌素释放蛋白的共表达[167];丝裂霉素诱导的细菌素释放蛋白和在培养基中添加甘氨酸[168]以及与kil基因共表达而进行膜穿透[169]。但通常情况下,外泌蛋白质的产量是中等的。
有文献报道[170],在大肠杆菌表达系统中,金黄色葡萄球菌A蛋白的信号肽能引导带有E结构域的A蛋白片段或融合产物从细胞质外泌到培养基中,蛋白的外泌表达发生于细胞生长后期。但所用的启动子为A蛋白自身的启动子,该启动子在大肠杆菌中为非可控性的组成性表达,且强度较弱。如果能利用可控的强启动子进行A蛋白信号肽引导的基因表达,则有望在蛋白质外泌方面有所突破。目前我们正在进行这方面的尝试,且已经取得初步成效。
五、密码子的使用
原核和真核生物的基因对同义密码子的使用均表现非随机性[171,172]。对E.coli中密码子的使用频率进行系统分析得到以下结论[173]:(1)对于绝大多数的简并密码子中的一个或两个具有偏好;(2)某些密码子对所有不同的基因都是最常用的,无论蛋白质的含量多少,例如CCG是脯氨酸最常用的密码子;(3)高度表达的基因比低表达的基因表现更大程度的密码子偏好;(4)同义密码子的使用频率与相应的tRNA含量有高度相关性。这些结果暗示,富含E.coli不常用密码子(表1)的外源基因有可能在E.coli中得不到有效表达。
已经证明[174],微精氨酸tRNAArg(AGG/AGA)是多种哺乳动物基因在细菌中表达的限制因子。因为AGA和AGG在E.coli中不常用。如果共表达编码tRNAArg(AGG/AGA)的argU(dnaY)基因,就会高水平表达目的蛋白[174]。但利用同方法所进行的另外几项研究的结果却不一致[175,176]。其他的研究表明,通过用常用密码子替换稀有密码子或与“稀有”tRNA基因共表达可以提高外源基因在E.coli中的表达水平[177,178]。
许多研究者对有关密码子使用模式的进化意义和密码子使用效果的机制进行了研究,但至今尚未找到协调密码子的使用和转录本翻译的精确规则。似乎在转录本5’末端附近存在稀有密码子将会影响翻译效率。另外,基因5’编码区中GC含量似乎也影响其表达。这在人胸苷酸激酶(TS)中已经得到证明[179]。在不改变所编码蛋白的前提下,将TS cDNA的第三、四、五密码子的嘌呤碱基变成胸腺嘧啶,使得TS基因的表达占到E.coli中总蛋白量的25-30%。综上所述,有许多可变因素可能影响实验结果,如位置效应、稀有密码子的群集和mRNA的二级结构等等。
<center> </center>六、蛋白质的蛋白酶解
蛋白酶解是一个选择性的、高度调节的过程,该过程参与许多代谢活动。E.coli在细胞质、细胞外周质、内膜和外膜有许多蛋白酶[180,181]。这些蛋白酶参与宿主的代谢活动,如选择性地清除异常和错误折叠的蛋白。
到目前为止,蛋白酶解的机制尚未完全明了,但已有一些策略和方法以减少E.coli中异源蛋白的降解。虽然使得蛋白质不稳定的精确结构特点还不清楚,但是通过系统研究已经明确了一些蛋白不稳定的决定因素。蛋白酶解途径的“N-末端规则”在E.coli中能够发挥作用,即蛋白质的稳定性与其氨基端的残基有关[182]。在E.coli中,N-末端Arg、Lys、Leu、Phe、Tyr和Trp的半衰期为2分钟,而除脯氨酸外的其他氨基酸的半衰期均超过10小时。
有研究表明,在多肽的第二位带有较小侧链的氨基酸有利于甲硫氨酸氨肽酶催化的N-末端甲硫氨酸的去除,从而暴露出位于第二位的亮氨酸,使得该蛋白不稳定[183]。蛋白质的第二个决定因素是位于近氨基端的特异性内源赖氨酸残基[142,143]。该残基是多遍在蛋白链的受体,多遍在蛋白链在真核细胞中有利于遍在蛋白依赖的蛋白酶对蛋白质的降解。
有趣的是在一个多遍在蛋白中,它的两个决定簇可以位于不同的亚基上,却能靶向同一个蛋白进行加工[184]。氨基酸成分和蛋白质不稳定性的另一个关系体现在PEST假说中[185]。根据对短寿命真核蛋白的统计分析,蛋白质如果富含Pro、Glu、Ser和Thr的区域,且在该区域附近有某些特定的氨基酸,则该蛋白就会不稳定。这些PEST结构域的磷酸化导致钙的结合能力提高,从而利于钙依赖性蛋白酶对蛋白质的降解。这提示可以在缺乏PEST蛋白裂解系统的E.coli中表达PEST富含蛋白。
减少E.coli中重组蛋白裂解的策略有以下几种:(1)将蛋白质靶向细胞周质或培养基[145,186];(2)在较低的温度下培养细菌[187];(3)选用蛋白酶缺陷的菌株[188];(4)构建N-末端或C-末端融合蛋白[186];(5)将目的基因多拷贝串联[188];(6)与分子伴侣共表达[189];(7)与T4 pin基因共表达[190];(8)替换特定的氨基酸残基以消除蛋白酶裂解位点[191];(9)改善蛋白质的亲水性[192];(10)优化培养条件[193]。
七、融合蛋白表达
在E.coli中表达外源蛋白,尤其是真核蛋白时,蛋白质的稳定性是经常遇到的问题。最近几年,众多巧妙的蛋白质——融合系统的发展,为E.coli中高效表达和纯化重组蛋白提供了极大方便。融合表达具有多方面的优点:如防止包涵体的形成,促进蛋白质的正确折叠,限制蛋白酶解和利于纯化[159,194]。
Uhlen和其同事[195]利用葡萄球菌A蛋白和合成的结构域(Z)开发了一种多功能的融合伴侣,除了能够作为纯化标记外,A蛋白组分还作为一种可溶性伴侣促进蛋白质的折叠,A蛋白信号肽的存在可使表达蛋白分泌到培养基中。另一个融合伴侣是链球菌G蛋白(SPG),它是一种细菌胞壁蛋白,在其氨基端具有分离的白蛋白结合区,在OH端具有免疫球蛋白IgG结合区[196]。最小的白蛋白结合区由来源于SPG的46个氨基酸残基组成,作为亲和纯化标记纯化cDNA编码的蛋白。如果将A蛋白和SPG结构域联合组成三联融合蛋白,则为纯化提供了更为广泛的选择,可以更进一步防止蛋白酶解。SPG-白蛋白的一个重要应用是其能够稳定哺乳动物外周循环中的短寿命蛋白,这一效应是通过SPG结构域与一种长寿命蛋白——血清白蛋白的结合来介导的[197]。
最近又建立了一种更为复杂和巧妙的亲和系统[198]。这种多元系统利用了七种不同的亲和标记,从而允许使用多种结合和洗脱条件,为重组蛋白的生产、检测和纯化提供了一个有力的工具。
使用基因融合表达系统在E.coli中表达外源基因已经越来越受欢迎。这在很大程度上归因于融合系统能够产生大量的可溶性的融合蛋白。谷胱甘肽S-转移酶(GST)、麦芽糖结合蛋白(MBP)以及硫氧还蛋白(Trx)均已经被证实能非常成功地生产正确折叠、有生物活性的蛋白质,能明显提高在E.coli细胞质中产生的融合蛋白的可溶性,并能抑制包涵体的形成[159,194]。
其中每一种都备有方便的纯化方法,可将融合蛋白与细胞污染物分开。已经建立了多种对融合蛋白进行位点特异性裂解的方法,方法的选择通常由特定蛋白的组成、序列及物理性质决定[199]。可采用诸如溴化氰(Met↓)、羟胺(Asn↓Gly)、等试剂或低pH(Asp↓Pro)来进行融合蛋白的化学裂解。
化学裂解的方法较便宜而且有效,甚至常常可以在变性的条件下裂解非变性不能溶解的蛋白质。但有时目的蛋白中存在裂解位点,或因发生副反应而导致对蛋白质进行不必要的修饰,从而阻碍了它们的应用。作为一个备选方案,酶解的方法相对来说其反映条件较温和,更重要的是,普遍用于此用途的蛋白酶都具有高度的特异性。其中常用的酶有:Xa因子、凝血酶、肠激酶、凝乳酶和胶原酶。所有这些酶都具有较长的底物识别序列,从而降低了蛋白质中其他无关部位发生断裂的可能性。在上述提及的各种酶中,Xa因子和肠激酶应用最多,因为它们切割各自的识别序列的羧基端,使带有天然氨基酸的被融合部分得以释放。
八、分子伴侣
目前已经达成共识,有效的蛋白质翻译后折叠、多肽装配成寡聚体结构以及蛋白质的转位都是由一种被称为分子伴侣的专职蛋白来介导的[200]。原核生物的核酮糖-1,5-二磷酸羧化酶在E.coli中的有效合成和装配需要GroES和GroEL蛋白的证据,使得利用分子伴侣在E.coli中进行基因高效表达成为近来研究的热点[201]。但是,利用分子伴侣所得到的实验结果并不一致,而且迄今为止,伴侣分子的共表达对基因表达的影响似乎都具有蛋白质特异性[202]。目前尚不清楚在基因过度表达的情况下,分子伴侣的体内水平是否受到限制。
正常情况下,蛋白质的折叠最终达到一种热力学的稳定状态。特别不稳定的蛋白即使在伴侣分子存在的情况下,或许也不能正确折叠。因此,多肽链的截断、多亚基蛋白复合物单个结构域的产生、缺乏维持蛋白质正常结构的二硫键的形成以及缺乏翻译后的修饰如糖基化等,都将不可能达到热力学的稳定状态。现在已经明白不同类型的伴侣分子正常情况下是协同发挥作用的[203]。因此,只过度表达单一的伴侣分子可能不太有效。在某些情况下,共表达与靶蛋白来源相同的伴侣分子可能是必要的。
还有一个需要考虑的变量是培养温度。例如,在30℃时GroES-GroEL共表达能够提高β-半乳糖苷酶的产量,而在37℃或42℃则不能[204]。最后,伴侣分子的共表达有可能导致表型的改变如细菌丝状生长,这有可能对细菌的生存和蛋白质的产生不利[205]。
最近有报道表明,将人或鼠的蛋白质二硫键异构酶(PDI)与靶基因共表达,能提高在E.coli细胞质中正确折叠蛋白质的产量[206,207]。E.coli细胞质中二硫键的形成是由维持氧化还原电势的一组蛋白质来促进的[208]。有人认为DsbA(一种可溶性的细胞外周质蛋白)直接催化蛋白质中二硫键的形成,而DsbB(一种内膜蛋白)则参与DsbA的再氧化。真核生物的PDI能够补充dsbA缺失突变株的表型,但其功能在dsbB突变株中完全丧失。另外,通过额外添加谷胱甘肽可以提高PDI增强靶蛋白产生的能力。这些证据表明,PDI有赖于细菌氧还蛋白来完成自身的再氧化[207]。
九、培养条件
E.coli中的蛋白质产量可以通过高细胞密度培养系统而获得显著提高。高细胞密度培养系统可以分成三类:分批培养、补料分批培养和连续培养。这些方法能获得超过100g/升的细胞浓度,从而获得廉价的重组蛋白。有关大规模培养系统的资料已有详细的综述[193,209]。培养基的组成需要仔细地计算和监控,因为这对细胞和蛋白质的产生具有重要的代谢效应。
例如,不同mRNA的翻译可以因温度和培养基的变化而受到不同程度的影响[210]。营养成分和培养参数如pH、温度和其他参数都会影响蛋白酶的活性、分泌和产量[211]。已经证明对培养基的特殊操作能明显提高蛋白质释放到培养基中。例如,在培养基中添加甘氨酸能增强外周质蛋白释放到培养基中,且不引起明显的细菌裂解[212,213]。同样,在山梨糖醇和甘氨酰甜菜碱存在的渗透压力下培养细菌,可以使可溶性的活性蛋白产量提高多达400倍[139]。
高细胞密度培养系统也有其自身的缺陷。这些缺陷包括在高细胞密度情况下,溶解氧的量有限;二氧化碳水平能够降低生长速度、刺激乙酸形成降低发酵罐的混合效率和产热等。有关这些问题的解决方案已经有详细的介绍[193]。利用高细胞密度培养系统生产重组蛋白质的一个主要问题是乙酸的积累,这种亲脂性成分对细胞的生长是有害的[193]。虽然有多种解决这一问题的方案,但是均各有缺点。近来这一问题通过将来自B.subtilis编码醋酸盐合成酶的alsS基因导入E.coli细胞中得以解决[141]。该酶催化丙酮酸转化为非酸性和低毒性的副产品。乙酸积累的减少极大地改善了重组蛋白的产生。另外,其他酶缺陷的E.coli突变株也已经建立,这些突变株产生较少的乙酸,从而提高了人重组蛋白的表达水平[214]。
总结
一种高效的原核表达载体需要包括一个强大并且可以严紧调节的启动子;一位于翻译起始密码子5’端大约9bp的SD序列;位于目的基因3’末端的一个高效转录终止子。除此之外,载体还需要一个复制起点,筛选标记和利于对启动子活性进行严紧调节的基因。这种调节元件可以插入载体自身,也可以插入宿主的染色体。其他的元件包括转录和翻译增强子等。这些元件的作用往往具有基因特异性,因此要根据不同的情况加以取舍。我们在综合国内外研究的基础上,对有可能影响E.coli中蛋白质高效表达的多种因素进行了总结。尽管目前在E.coli中表达外源基因方面已经有许多重大进展,但仍有许多问题亟待解决。归纳起来主要有以下几点:
1. 借助于细胞的分子伴侣机制来提高正确折叠的蛋白质的产量。或许可以通过共表达多种分子伴侣编码基因和通过激细胞内众多不同的分子伴侣的方法来实现。
2.在E.coli中表达真核膜蛋白和多亚基蛋白质复合物的难题上有待解决。
3. 蛋白质分泌到培养基中的真正和有效机制需要明了。目前已经有多种体系能够是重组蛋白质分泌到培养基中。其中有些是利用信号肽、融合伴侣和具有穿透能力的因子,这种因子能够引起外膜的破坏和有限的渗漏。而另外的策略是利用已经存在的分泌通道,来保证更高特异性的分泌。有关这方面的工作需要对E.coli中众多分泌通道的更多了解。
4. 赋予原核细胞诸如真核细胞那样进行翻译后修饰的能力。例如糖基化、磷酸化、乙酰化和酰氨化等。可以通过将合适的真核细胞酶类转入E.coli中而达到这些目的。
总之,E.coli作为一种原核表达系统具有多方面的优点,加之在许多技术方面的众多重大进展。E.coli仍然是基础研究和商业生产重组蛋白质的强有力工具。
参考文献
1.吴加金 生物工程学报12(增刊),1996,92-96
2.Rober C.Hockney TIBTECH.1994,12:456-464
3.Georgion G.in Protein Engineering: Principles and Practice(Cleland,J.I.,and Craik,C.S.,eds).1996,101-127,Wiley-Liss
4.张智清,姚立红,侯云德,病毒学报,1990,6:102
5.张智清,张颖,病毒学报,1993,9:136-142
6.金冬雁,徐荣辉,周 圆等,中国科学(B辑),1992,22(9):951-957
7.薛水星,吴淑华,韩 峰等,生物工程学报12(增刊),1996,62-66
8.Gerihard Hannig,Savvas C.Makrides.TIBTECH.1998,16:54-60
9.Galas D J,Eggert M,Waerman M S.J Mol Biol.1985,186:117-128
10.Harley C B,Reynolds R P.Nucleic Acids Res.1987,15:2343-2361
11.Hawley D K,McClure W R.Nucleic Acids Res.1983,11:2237-2255
12.Yansura D G,Henner D J.Methods Enzymol.1990,185:54-60
13.Huttenhofer A,H F Noller L.EMBO J.1994,13:38920-3901
14.Shine J,Dalgarno L.Proc Natl Acad Sci USA.1974,71:1342-1346
15.Shine J,Dalgarno L.Nature.1975,254:34-38
16.de Smit M H,van Duin J.J Mol Biol.1994,235:173-184
17.Chen H Y,Bjerknes M,Kumar R,Jay E.Nucleic Acids Res.1994,22;4953-4957
18.Gold L,Stormo G D.Methods Enzymol.1990,185:89-93
19.Dreyfus M.J Mol Biol.1988,204:79-94
20.Rosenberg M,Court D.Annu Rev Genet.1979,13:319-353
21.Belasco J G,Higgins C F.Gene.1988,72:15-23
22.Berkow R.The Merck manual of diagnosis and therapy,16th ed.,p24-30.
23.Morino T,Morita M.Appl Microbiol Biotechnol.1988,28:170-175
24.Nordstrom K,Uhlin B E.Bio/Technology.1992,10:661-666
25.Vasquez J R,Evnin L B.J Cell Biochem.1989,39:265-276
26.Minas W,Bailey J E.Biotechnol Prog.1995,11:403-411
27.Brown W C,Campbell J L.Gene.1993,127:99-103
28.Doherty A J,Connolly B A,Worral A F.Gene.1993,136:337-340
29.William K L,Emslie K R,Slade M B.Curr Opin Biotechnol.1995,6:538-542
30.Yike L,Y Zhang,Ye J,Dearborn D G.Protein Expression Purif.1996,7:45-50
31.O'Connor C D,Timmis K N.J Bacteriol.1987,169:4457-4462
32.Bentley W E,Mirjalili N,Andersen D C.Biotechnol Bioeng.1990,35:668-681
33.Chen W,Kallio P T,Balley J E.Biotechnol Prog.1995,11;397-492
34.Lanzer M,Bujard H.Proc Natl Acad Sci USA.1988,85:8973-8977
35.de Boer H A,Comstock L J,Vasser M.Proc Natl Acad Sci USA.1983,80:21-25
36.Brosius J,Erfle M,Storella J.J Biol Chem.1985,260:3539-3541
37.Flgge J,Wright C,Collins C J.Cell.1988,52:713-722
38.Bukrinsky M I,Barsov E V,Shilov A A.Gene.1988,70:415-417
39.Adari H,Andrew B,Ford P J.DNA Cell Biol.1995,14:945-950
40.Andews B,Adari H,Hannig G.Gene.1996,182:101-109
41.Ward G A,Stover C K,Moss B.Proc Natl Acad Sci USA.1995,92:6773-6777
42.Yabuta M,Onai-Miura S,Ohsuye K.J Biotechnol.1995,39:67-73
43.Xue G P,Johnson J S,Smyth D J,Appl Microbiol Biotechnol.1996,45:120-126
44.Giladi H,Goldenberg D,Koby S.Proc Natl Acad Sci USA.1995,92:2184-2188
45.Freundlich M,Ramanl N,Mathew E.Mol Microbiol.1992,6:2557-2563
46.Friedman D L.Cell.1988,55:545-554
47.Goldstein J,Pollitt N S,Inouye M.Proc Natl Acad Sci USA.1990,87:283-287
48.Oppenheim A B et al Internal patent application WO 96/03521 1993
49.Chou CH,Aristidou A A,Meng S Y.Biotechnol Bioeng.1995,47:186-192
50.Tolentino G J,Meng S Y,San K Y.Biotechnol Lett.1992,14:157-162
51.San K Y,Bennet G N,Chou CH.Ann N Y Acad Sci.1994,721:268-276
52.Deuschle U,Kammerer W,Gentz R.EMBO J.1986,5:2987-2994
53.Gaal T,Barkei J,Dickson R R.J Bacteriol.1989,171:4852-4861
54.Gourse R L,de Boer H A,Nomura M.Cell.1986,44:197-205
55.Hsu L M,Glannini J K,Leung T W C.Biochemistry.1991,30;813-822
56.Josaitis C A,Gaal T,Gourse R L.Biochem Biophys Acta.1990,1050:307-311
57.Rao L,Ross W et al J Mol Biol.1994,235:1421-1435
58.Ross W,Gosaink K K et al Science.1993,262:1407-1413
59.Gafuy R,Cohen S,Nachaliel N.J Mol Biol.1994,243;152-156
60.Josaitis C A,Gaal T,Gourse R L.Proc Natl Acad Sci USA.1995,92;117-1121
61.Brosius J,Holy A.Proc Natl Acad Sci USA.1984,81:6929-6933
62.Chalmberlin M J.Harvey Lect.1994,88:1-21
63.Forsberg G J.Protein Chem.1992,11:201-211
64.Adhya S,Gottesman M.Cell.1982,29:939-944
65.Nishihara T,Nohno T.Gene.1994,145:145-146
66.Hayashi M N,Hayashi M.Nucleic Acids Res.1985,13:5937-5948
67.Newbury S F,Smith NH.Cell.1987,48:297-310
68.Brosuius J,Ullrich A,Rake M A.Plasmid.1981,6:112-118
69.Landicak R,Turnbough C L.Transcription Attenuation.1996,p1263-1286
70.Berg K L,Squires C,Squires C L.J Mol Biol.1989,209;345-358
71.Li S C,Squires C L,Squres C.Cell.1984,38:851-860
72.Studier F W,Moffatt B A.J Mol Biol.1986,189:113-130
73.Tabor S,Richardson C C.Proc Natl Acad Sci USA.1985,82:1074-1078
74.Mertens N,Remaut E,Flers W.Bio/Technology.1995,13:175-179
75.Brosius J.Methods Enzymol.1992,216:469-483
76.Taylor A,Brown D P,Kadam S.Appl Microbiol Biotechnol.1992,37:205-210
77.Muller-Hill B,Crapo L,Gllbert W.Proc Natl Acad Sci USA.1968,59:1259-1264
78.Warne S R,Thomas C M,Nugen M E.Gene.1986,46:103-112
79.Mertens N,Remaut E,Flers W.Gene.1995,164:9-15
80.Thomas C D.Gene.1993,136:237-242
81.Chen W,Kallio P T,Bailey J E.Gene.1993,130:15-22
82.Sagawa H,Ohshima A,Kato I.Gene.1996,168:37-41
83.Backman K,O'Connor M J,Maruya A.Bio/Technology.1984,2:1045-1049
84.Balakrishnan R,Bolten B,Backman K C.Gene.1994,138:101-104
85.Stark M J R.Gene.1987,51:255-267
86.Hasan N,Szybalski W.Gene.1995,163:35-40
87.Kobayashi M,Nagata K,Ishihama A.Nucleic Acids Res.1990,18:7367-7372
88.Gren E J.Biochimie.1984,66:1-29
89.Gualerzi CO,Pon C L.Biochemistry.1990,29;5881-5889
90.Gold L.Annu Rev Biochem.1988,57:199-233
91.Barrick D,Villanueba K,Childs J.Nucleic Acids Res.1994,22:1287-1295
92.de Smit M H,van Duin J.J Mol Biol.1994,244:144-150
93.de Smit M H,van Duin J.Proc Natl Acad Sci USA.1990,87:7668-7672
94.Ringquist S,L Goild M et al Mol Microbiol.1992,6:1219-1229
95.Hall M N,Gabay J,Schwartz M.Nature.1982,295:616-618
96.Hartz D,McPheeters D S,Gold L.J Mol Biol.1991,218:83-97
97.Gheysen D,Iserentant D,Derom C.Gene.1982,17;55-63
98.Chen H Y,Pomeroy-Cloney L,Bjerknes M.J Mol Biol.1994,240:20-27
99.Coleman J,Inouye M,Nakamura K.J Mol Biol.1985,181:130-143
100.Schumperli D,Cell.1982,30:865-871
101.Boni I V,Isaeva D M,Tzareva N V.Nucleic Res.1991,19:155-162
102.Olins PO,Rangwala SH.Gene.1988,73:227-235
103.Olins PO,Rangwala SH.J Biol Chem.1989,264:16973-16976
104.McCarthy J E G,Schairer H U,Sebald W.EMBO J.1985,4:519-526
105.McCarthy J E G,Sebald W,Gross G.Gene.1986,41;201-206
106.Zhang J,Deutscher M P.J Biol Chem.1989,264:18228-18233
107.Tzareva N V,Makhno V I,Boni I V.FEBS Lett.1994,337:189-194
108.Sprengart ml,Fatscher H P,Fuchs E.Nucleic Acids Res.1990,18:1719-1723
109.Sprengart ml,Porter A G,Remaut E.EMBO J.1996,15:665-674
110.Belasco J G,Control of messenger RNA stability.Academic Press,Inc.,San Diego,Calif 1993,p3-12
111.Chen C Y,Belasco J G.J Bacteriol.1990,172:4578-4586
112.Bechhofer D H.Control of messenger RNA stability.Academic Press,Inc.,San Diego,Calif 1993,p3-12
113.Higgins C F,Causton H C.Control of messenger RNA stability.Academic Press,Inc.,San Diego,Calif 1993,p13-20
114.Duvoisin R M,Belin D,Krisch H M.Gene.1986,45:193-201
115.Bechhofer D H,Dubnau D.Proc Natl Acad Sci USA.1987,84:498-502
116.Yamamoto T,Imamoto F.J Mol Biol.1975,92:289-309
117.Belasco J G,Nilsson G,Coben S N.Cell.1986,46:245-251
118.Emory S A,Bouvet P,Belasco J G.Genes Dev.1992,6:135-148
119.Wong H C,Chang S.Proc Natl Acad Sci USA.1986,83:3233-3237
120.Donovan W P,Kushner S R.Nucleic Acids Res.1983,11:265-275
121.Donovan W P,Kushner S R.Proc Natl Acad Sci USA.1986,83:120-124
122.Craigen W J,Lee C C,Caskey C T.Mol Microbiol.1990,4:861-865
123.Scolnik E,Tompkin R.Proc Natl Acad Sci USA.1968,61:768-774
124.Grentzmann G.Proc Natl Acad Sci USA.1994,91;5848-5852
125.Sharp P M,Bulmer M.Gene.1988,63:141-145
126.Tate W P,Brown C M.Biochemistry.1992,31:2443-2450
127.Poole E S,Brown C M,Tate W P.EMBO J.1995,14:151-158
128.Mottagul-Tabar S,Bjornsson A,Isaksson L A.EMBO J.1994,13:249-257
129.Bjornsson A,Mottagui-Tabar S,Isaksson L A.EMBO J.1996,15:1696-1704
130.Wilkinson D L,Harrison R G.Bio/Technology.1991,9:443-448
131.Schein CH.Curr Opin Biotechnol.1991,2:746-750
132.Cabilly S.Gene.1989,85:553-557
133.Kenealy W R,Gray J E,et al Dev Ind Microbiolo.1987,28:45-52
134.Dale G E,Broger C,Langen H.Protein Eng.1994,7;933-939
135.Amrein K E,Takacs B,Stieger M.Proc Natl Acad Sci USA.1995,92:1048-1052
136.Batistoni A,Carri M T,Rotilio G.FEBS Lett.1993,322:6-9
137.Blum P,Ory J,Krska J.J Bacteriol.1992,174:7436-7444
138.LaVallie E R,Bio/Technlogy.1993,11:187-193
139.Blackwell J R,Horgan R.FEBS Lett.1991,295:10-12
140.Sugimoto S,Yokoo Y.Biotechnol Lett.1991,13:385-388
141.Aristidou A A,San K Y,Bennett G N.Biotechnol Bioeng 1994,44:944-951
142.Bachmair A,Finley D,Varshavsky A.Science.1986,234:179-186
143.Bachmair A,Varshavsky A.Cell.1989,56:1019-1032
144.Swamy K H S,A L Goldberg.J Bacteriol.1982,149:1027-1033
145.Talmadge K,Gilbert W.Proc Natl Acad Sci USA.1982,79:1830-1833
146.Michaelis S,Beckwith J.Annu Rev Microbiol.1982,36:435-465
147.Schatz G,Dobberstein B.Science.1996,271:1519-1526
148.Schatz P J,Beckwith J.Annu Rev Genet.1990,24:215-248
149.Deneffe P,Kovarik S,Clora T.Gene.1989,85:499-510
150.Ghrayeb J,Kimura H,Takahara M.EMBO J.1984,3;2437-2442
151.Johnson D L,Middleeton S A.Protein Expression Purif.1996,7:104-113
152.Hoffman C S,Wright A.Proc Natl Acad Sci USA.1985,82:5107-5111
153.Hogset A,Blingsmo O R.J Biol Chem.1990,265:7338-7344
154.Schein CH,Boix E.Biochem J.1992,283:137-144
155.Gray G L,baldridge J S.Gene.1985,39;247-254
156.Pluckthun A.Immunol Rev.1992,130:151-188
157.van Dijl J M,de Jong A.Mol Gen Genet.1991,227:40-48
158.Snyder W B,Silhavy T J.J Bacteriol.1992,174:5661-5668
159.Makrides S C.Microbiol Rev.1996,60:512-538
160.Pugsley A P.Microbiol Rev.1993,57:50-108
161.Stader JA,Silhavy T J.Methods Enzymol.1990,185:166-187
162.Blight M A,Chervaux C,Holland I B.Curr Opin Biotechnol.1994,5:468-474
163.Holland L B,Steipe B.Methods Enzymol.1990,182:132-143
164.Better M,Chang C P,Robinson R R.Science.1988,240:1041-1043
165.Ko J H,Park D K.Biotechnol Lett.1995,17:1019-1024
166.Abrahmsen L,Moks T,Nilsson B.Nucleic Acids Res.1986,14;7487-7500
167.Hsiung H M,Cantrell A.bio/Technology.1989,7:267-271
168.Yu P,Aristiou A A,San K Y.Biotechnol Lett.1991,13:311-316
169.Kat℃,Kudo T.et al Gene.1987,54:197-202
170.Abrahmsen L,Moks T,Nilsson B.Nucleic Acids Res.1985,4:3901-3906
171.Gouy M,Gautier C.Nucleic Acids Res.1982,10:7055-7074
172.Gutman G A,Hatfield G W.Proc Natl Acad Sci USA.1989,86:3699-3703
173.de Boer H A,Kastelein R A.Biased codon usage.1986,p225-285
174.Brinkmann U,Mattes R E,Buckel P.Gene.1989,85:109-114
175.Chen G F T,Inouye M.Genes Dev.1994,8:2641-2652
176.Chen K S,Peters T C,Walker J R.J Bacteriol.1990,172:2504-2510
177.Buell G,Schulz M F,Selzer G.Nucleic Res.1985,13:1923-1938
178.Devli P E,Drummond R J,Toy P.Gene.1988,65:13-22S
179.Pedersen-Lane J,Maley G F,Chu E et al Protein Expres Purif 1997,10:256-262
180.Goldberg A L.Eur J Biochem.1992,203:9-23
181.Gottesman S,Maurizi M R.Microbiol Rev.1992,56:592-621
182.Tobias J W,Shrader T E,Rocap G et al Science.1991,254:1374-1377
183.Hirel PH,Dessen P,Fayat G.Proc Natl Acad Sci USA.1989,868247-8251
184.Johnson E S,Gonda D K,Varshavsky A.Nature.1990,346:287-291
185.Rogers S,Wells R,Ptashne M.Science.1986,234:364-368
186.Hammarberg B,Nygren P A,Holmgren E.Proc Natl Acad Sci USA .1989,864367-4371
187.Chesshyre J A,Hipkiss A R.Appl Microbiol Biotechnol.1989,31:158-162
188.Shen SH.Proc Natl Acad Sci USA.1984,81:4627-4631
189.Sato K,Sato M H,Yoshida M et al Biochem Biophys Res Commun.1994,202:258-264
190.Simon L D,Randolph B,Irwin N et al Proc Natl Acad Sci USA.1983,80:2059-2062
191.Hellebust H,Murby M.Bio/Technology.1989,7:165-168
192.Murby M,Samuelsson E,Nguyen T N.Eur J Biochem.1995,230:38-44
193.Lee S Y,Trends Biotechnol.1996,14:98-105
194.Lavallie E R,McCoy J M.Curr Opin Biotechnol.1995,6:501-506
195.Moks T,Abrahmsen L,Uhlen M.Biochemistry.1987,26:5239-5244
196.Falkenberg C,Bjorck L,Akerstrom B.Biochemistry.1992,31:1451-1457
197.Nygren P A,Uhlen M,Flodby E.Vaccine.1991,91:363-368
198.Nilsson J,Larsson M,Uhlen M.Eur J Biochem.1994,224:103-108
199.Lee N,Cozzikorto J,Wainwright N.Nucleic Acids Res.1984.12:77-86
200.Becker J,Craig E A.Eur J Biochem.1994,219:11-23
201.Cole P A.Structure.1996,4:239-242
202.Wall J G,Pluckthun A.Curr Opin Biotechnol.1995,6:507-516
203.Buchner J.FASEB J.1996,10:10-19
204.Lee S Y,Olins PO.J Biol Chem.1992,267:2849-2852
205.Georgiou,G.,P.Valax.Curr.Opin.Biotechnol.1996,7:190-197
206.Humphreys D P,Weir N.J Biol Chem.1995,270:28210-28215
207.Ostermeier M,De Sutter K,Georgiou G.J Biol Chem.1996,271:10616-10622
208.Bardwell J C A,Mol Microbiol,1994,14:199-205
209.Yamane T,Shimizu S.Adv Biochem Eng.1984,30:147-194
210.Jacques N,Guillerez J,Dreyfus M.J Mol Biol.1992,226:597-608
211.Enfors SO.Trends Biotechnol.1992,10:310-315
212.Ariga O,Andoli Y,Fugishita Y.J Ferment Bioeng.1991,71:397-402
213.Aristidou A A,Yu P,San K Y.Biotechnol Lett.1993,15:331-336
214.Bauer K A,Ben-Bassat A,Dawson M.Appl Environ Microbiol.1990,56:1296-1302