基因表达之RT-PCR之我见
互联网
在论坛上看到不停地有人在问RT-PCR的问题,实际上在以前的帖子中各位香主和eeflying(我为什么总是提他们?因为还是很佩服的哦,生怕自己说错了,被他们指出来哦)都谈到了很多,鄙人也充大头答了一些问题。
但是还是有各种问题,由于的基因表达还有点实战经验(但也技止此而),所以想写点东西给大家参考,计划把包括RNA酶保护分析,northernblot,原位杂交,半定量RT-PCR和定量RT-PCR都写了,但是实在时间不够,我从来不写综述的,写这么多还是头一次,声明一下,全部文字均为原创(当然如有雷同,那就是纯属巧合了),各位如要引用,请来信告知,最好著注明来自丁香园,实际上国外现在引用网站内容已成为常见的事情了,nature和science都有引用网站的,以后大家的文章的引用的是丁香园,该有多爽。
本文不谈具体protocol,是因为各种书籍和kit说明书上都有,主要说一些原则,而且大部分是失败和成功的经验,还有我们小组的讨论结果,以及各种书里七零八落看的内容,泣血力作,希望对大家有用。
方法:很多很多,我们常用的也就是RT-PCR和northern(实际上我也只知道他们,丢人啊),但是RNA酶保护分析在国外很常用,也有半定量的功能,一次性试剂盒还能以此将一个表达簇一起分析,例如NFkB的转录激活谱中的8个常见基因。
至于dot blot和原位杂交之流则实属于吃力不讨好,不知所云,骗人玩玩的啦,dot blot的最大贡献是发展到了反向dot blot的顶峰――曾经无限时髦的基因芯片,而原位杂交的放大效应和它的假阳性使它的定量(半定量,应该)根本算不上定量(至于图象分析一般是给自己“找一个理由先”),用它定位和用蛋白定位的意义不可同日而语,又难做,只有新发现的基因可以在没有得到蛋白和抗体的情况下进行组织或者细胞定位。
愚见采用从RNA定量的方法似不妥,不要说PCR的本身的敏感度,即便是在反转录就有差异,到了cDNA的分装和用于模板的取样,这些都不能保证准确,当然在反转录阶段我们也经常控制在1或5ug,但这是为了再将来加样的时候保证大致差不多,而且尽可能地利用反转录酶,而且RNA定量本身即使用电泳也不是那么准,更不要说风光光度计,这里说一下,我曾经看到有人说用分光光度计定量,这也不是很准确,分光光度计只是掌握大概,当然用于反转录足够了,但是用于rt-pcr的定量这是不正确的,很不准的,RNA也最好至少要用电泳,一方面定量,另一方面可以看看完整性。至于用分光光度计比较不同标本之间就更不准了。
略。什么手套、DEPC、干烤、沉淀等等书里和各个帖子里都有。一般制备总RNA 即可,有时需要制备mRNA,个人认为初学者还是选择Trizol,大量制备采用传统的方法自己配,实际上和trozol一样的,有时效果还好,trizaol也就是混一混,但是优点在于质量保证,使用方便,效率高,根据不同组织用trizol一般可以提到全部RNA的50%(别小看,这已经很高了)以上,有些可以到达80%。mRNA用Qiagen的柱子,别去自己做Oligo(dT)柱,太麻烦。
是否用DNAseI根据基因的特点和引物的设计,能够跨内含子的就不需要处理,处理还是很危险的哦。像这样的微量和也用不了多少次的实验,并且如果牵涉到样品是无价之宝的时候,因该选好的进口试剂。废话几句,在另一个论坛上看到一贴谈到RNA的贮存,因为没有登陆在那里也不能说,可能对大家有启发,在这里说一下,实际上主要是温度,至于放在TRIZOL中是不可取的,更不要说在胍里了,只-20是不够的,至少-80,液氮最保险,想想,在低温里,即使加上些RNA酶它也降解不了哇!qiagen出了一种easy产品可用于保存标本,但在常温也只是1天,所以低温才是首要的,抽的时候也是这样。
常规,取样尽量一致,如上所述,不是为了定量,是为了半定量PCR时图形的好看。选择逆转录酶根据实验需要,少量的可用Promega的一种1ug的酶,多的我们用invitrgen的5ug的II,当然有人说Promega和Roche的也不错,用过,的确可以,要不Invitrogen不像当初Gibco时那么牛了,也降价打折了。反转录成功后就不必太小心了,放在那里都可以,想想你还曾经72度灭活呢,是不是?
要知道,杂合双链比DNA双链还稳定的。另外把回答的一封信粘上:一般来说要扩增从反转录到PCR全过程的长达2kb以上的片断是相当困难的事(我瞎掰的),很多长基因是通过随机引物库得到的而不是通过oligo (Td)得到的,不要说反转录,即使是PCR也是很困难的,不信可以从质粒里试试,至于反转录,就更是天方夜谭了,除了酶的效率等,还有RNA的二级结构等因素,当然,霸王硬上弓未尝不可,这就要一些大公司的特殊的名贵的效果也未必好的酶了,Roche和Invitrogen都有的.听天由命吧,不过,即使××,未必成功。
重点,为什么是第4条? 首先说明一下,半定量PCR,我是指基于凝胶成像分析的,定量PCR指实时PCR。
引物:除了常规引物设计原则,这在各种书籍上都有的,在园里各位主任像yong啊eeflying啊什么的也说了很多了,eeflying的有图的那一贴偶是看不懂得,什么时候解释解释。此处不谈。我看过最好的是郑仲承写的一篇综述,丁香圆贴过,经典啊,这一辈子是赶不上了,自己真是井底之蛙,还在狂叫不止。对于RT-PCR重要的是引物的位置,有两种方法,一种是上下游引物设计在跨内含子的两个外显子的3’端和下一个外显子的5’端,这样不会在基因组上扩出来。
第二种是我最喜欢的,设计在两个离得远的外显子上,这样从基因组和cDNA上得到得不一样,可以一引两用,^_^。以上原则对单外显子基因不使用,这是最好选择DNAaseI处理。另外,我们有一个好习惯(嘿嘿,偶已经打了一把伞),就是把退火温度设计成一样的,这样每次做的时候不用“三查七对”,从冰箱里拿出就坐。另外3'最后一个碱基是很重要的。
不管多少样品,内参不作,等于说比较珠穆朗玛峰和泰山的高度一样,没有海拔,泰山从地面的高度和珠穆朗玛峰从青藏高原的高度没什么差别。PCR一定要同时作,但是电泳可以不一起跑,没有关系,记住,计算的是相对表达程度,再说一遍我的观点:
1、半定量和定量RT-PCR做的都是基因相对表达量,不是绝对表达量,除非你能准确知道来自多少细胞,但是细胞还有死的呢。
2、以电泳为基础的半定量RT-PCR本身是不可信的,作为实验的粗筛是可以的,但不能作为最终结果的,不过我的可以(苦胆吐出).3、半定量RT-PCR应该在两管中进行,除非内参基因和目的基因表达相同,长度一致,目的扩增区域的GC含量相似,或者实在穷的要省PCR管和Taq酶。
解释一下:
1、同一管中不是不可以,因为它有条件现同的优点,只要目的基因的扩增量和选择的不同循环数的内参照基因的最终扩增产量大致相同就很好,很拗口吧。但是一管中的竞争抑制实在是大问题,即使对照组一致了,咱做的是差异,总有不一致的,而且PCR的放大效应会把芝麻P成西瓜的哦。在不同管中重要的模板的平均分配,这不用多说了把。
2、相对和准确的问题,PCR的相对定量是因为引入了内参照,所以没有绝对定量一说,至于半定量和定量的区别不是相对和绝对的区别,而是准确和不准确的区别。一般经常搞错的定量半定量和绝对量相对量的概念,定量半定量是指检测准确度,而绝对量相对量是指基因在组织中的表达丰度,例如Northen虽然不是很准(Northern不是很好的金标准),主要是上样量的原因,不管使用28s18s定还是用SB GAPDH或者β-actin都是,但是Northern得出的是绝对组织丰度,而实时荧光定量RT-PCR虽然准确(定量)但是得出的是相对表达丰度,不管使用β-actin还是用18srRNA.至于常规的通过跑电泳的所谓”定量”RT-PCR那就远了去了。
关于选择什么作为内参照的问题,实际上没有定论,个人认为,18s优于actin,不要跟我说GAPDH,著名的GAPDH都可以说是肿瘤相关分子了,其它还有tubulin什么的,因该差不多,实际上actin虽然相对来说很稳定的表达量,但我想(是我想的,不一定对,哪位指教?),在细胞分裂的过程中,需要骨架,因此actin等本身在不同细胞,不同组织、不同时期是变化的,我想,姑妄听之。作为骨架蛋白和细胞的分裂发育是密切相关的,在不同状态表达是不同的,不同细胞等。
之所以用18s是因为相对而言核糖体RNA量相对稳定,因为它的功能是同整个基因谱有关的(负责装配),更重要的,(我想的),它在总RNA中占地比例高,所以更准确,就像你用某一种东西的数量去概括总量,因该选那种多的,说一座房子是由2000块砖造成的,比说用29根梁更准确吧,当然你用的是mRNA就另当别论了。
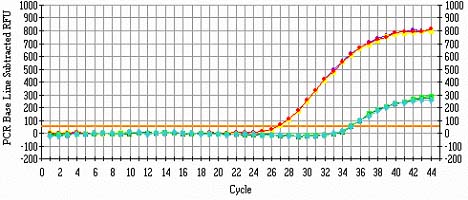
线性期和平台期:根据上面摸的结果,选择线性期的起始周围的循环数作为PCR指数,这和定量PCR中的原理是一样的,这相当于取线性期曲线的切线(是不是这样?)。线性期似乎因该是指数期(我想的),只是因为2的幂和倍数正好一样?不要为了后期的亮度取线性期的晚期,那样是不准的,文后复一张自备的定量RT-PCR的图参考就明白了。本来想把模板量单列的,在这里说了吧,模板量应该越多越好,当然是指在条件允许和不影响PCR体系的情况下,也不是指把20ul都加上,还要摸呢,对吧。
一般来说2ul要比1ul好,(唉,真知灼见都说出来了,(已经没有什么吐的啦吧))很多人会问,过了一个循环不久一样了么?不一样的,这和PCR本身的原理有关,什么原理我也不懂。就像到了线性期,很多人会说,再加酶,实际上不是的,再加酶和dNTP还有引物都没用的,其实这些本身就是过量的。
综合上面两点,说说下面一点:一般摸出来的结果是什么呢?个人认为,再使用1ugRNA反转录后用2-4ul为模板和使用5ug反转录用1-2ul为模板的情况下,一般目的基因很少超过30循环,著名的β-actin很少超过25循环,如果你看到有人用actin和sb GAPDH超过28-30循环的话,结果是不可信的,实际上即使超过25循环也是值得商榷的,肯定再RNA或者cDNA的步骤有问题,此时因该增加模板量,而不是增加循环数,即使目的基因要用30以上也要考虑一下。
对于想脾脏这样的组织,actin甚至可能要低于20循环,实际上在后文谈到的凝胶成像仪的检测范围之内,应该循环数越少越好。还是那句话,并不是在指数期就可以的。
关于凝胶成像分析:这是常人所未想的问题(晕倒一片),一般来说基于CCD的成像系统也有这个问题,这就是CDD本身的工作原理和软件的质量,有的CCD是重复扫描的,有的软件是只有256灰阶的,所以要注意上样量不要太多,当然也不要太少(以防看不到),否则会超出扫描CCD和软件的分析范围,这和PCR到线性期就没有什么不同了。
另外很多软件有手动调节,这样的软件使用时要保持每次框起来的范围的大小的一致性,不要造成太多的人为误差,号召大家不要任意修改数据。此外,紫外灯管的质量、光的强度、均一性和CCD距离、焦距、分辨率都要注意。如果有钱买冷CCD当然最好了,不过其实一般用不着。实在不行的,拍了照再扫描再用photoshop分析也未尝不可,但是那可是很不准的,中间不周太多,而且如果是热敏打印机很容易亮度饱和的,不一定会有灰阶。
据标准曲线检测基因啊病毒啊什么的绝对值。
为什么是准确的相对定量,我再废话一会again,是指你不知道你的模板来源是多少,所以才用GP内参,话说回来,及时你知道多少个细胞,还有细胞生死的问题对不?这就是为什么说northern是不很准确(没有定量PCR准确)的绝对定量,是因为northern的模板多,综合分光光度计和28s/18s更复合实际情况些(好像不能自圆其说,似乎也是相对定量),尤其是在不同组织分布的研究中更有效。