了解诱导多能干细胞无脑畸形的发病机制
发表在《分子脑》杂志上的一项新研究报告了利用诱导多能干细胞(iPSC)对人无脑畸形的发病机制进行了表征。在这篇博客中,庆应义塾大学医学院院长兼生理学教授冈野秀之博士向我们介绍了更多关于基于ipsc的无脑畸形模型的知识。无脑畸形是一种先天性的大脑畸形。
十年前,山中伸弥(Shinya Yamanaka)博士报道了获得诺贝尔奖的诱导多能干细胞(iPSCs)技术。除了它在体细胞重编程以成为多能性方面的意义之外,人们对ipsc技术也越来越感兴趣,包括应用于医学和开发新的治疗干预手段,使用基于细胞的再生医学和疾病建模。
我和我的同事一直在使用ipsc技术对神经和精神疾病进行疾病建模。我们已经从30多种神经和精神疾病中建立了iPSCs。这些疾病可分为儿童神经系统疾病、影响感觉器官的遗传性疾病、精神疾病和迟发性神经退行性疾病。在我们发表于《分子脑》的文章中,我们报道了严重的儿童神经系统疾病无脑畸形的特征。
无脑畸形是什么?
无脑畸形是最严重的先天性脑畸形之一,表现为大脑皮层分层紊乱。“无脑畸形”这个名字的意思是“光滑的大脑”,因为大脑表面由于旋转不足而显得光滑。
根据小鼠模型和人类遗传学的研究,已假定发育中的脑皮质的神经元迁移在无脑畸形中受到损害。在无脑畸形患者中,一些参与微管调控的基因,如LIS1、DCX、ARX和TUBA1A,都发生了突变。
因此,微管动力学的失调可能导致本病神经元迁移受损。然而,由于无脑畸形的罕见和早期的产后死亡,其分子和细胞病因学尚未得到实验研究。
我们做了什么?
为了克服这一困难,我们从患有TUBA1A错义突变的无脑畸形患者身上建立了iPSCs,编码了一种阿尔法微管蛋白亚型,在神经祖细胞和新生神经元中均有高表达。
我们对两例TUBA1A基因突变患者的间充质干细胞进行了特征分析。, R264C和N329S突变,这些突变可能分别影响-微管蛋白与Dcx蛋白的相互作用,减少-和-微管蛋白之间的氢键数量。
为了在体外描述可能与大脑皮层分层紊乱相关的表型,我们使用了我们之前开发的方法,神经胶质支持的神经突扩展方法。该方法可观察到神经祖细胞(或年轻神经元)的移位样运动;因此,我们希望我们的共培养系统可能在ipsc衍生的端脑神经元的神经元迁移中发挥作用。
我们发现了什么?
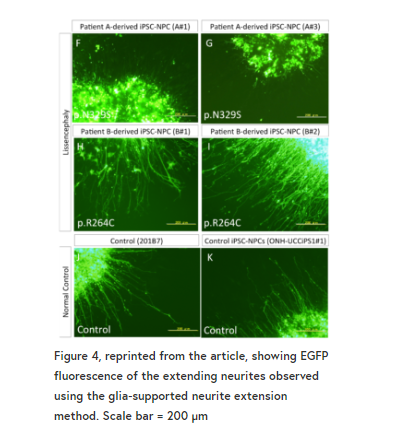
在神经胶质支持的神经突扩展实验中,我们发现从严重的无脑畸形患者来源的iPSCs分化而来的神经祖细胞的神经突扩展受到严重干扰。
术后5天的神经突延长量测定表明,iPSC-NPCs神经突延长来源于p。N329S TUBA1A突变被严重抑制(图4 F-G和L),这种神经伸展抑制在B患者中观察较少(图4 H-K),这与患者沿头尾轴的病理梯度一致。
这些数据表明,神经细胞从ipsc - npc与p。与对照组相比,N329S TUBA1A突变体的形态不成熟。我们认为这种表型与大脑发育的病理相对应,因为神经元细胞最初从npc分化,并通过各种类型的神经元突起(包括前导过程和基底过程)向皮质板迁移。
这是首次报道脑无裂畸形的神经突起扩展特征,可以认为脑无裂畸形-间充质干细胞为了解微管在脑无裂畸形发病机制中的作用以及人脑皮层发育机制提供了线索。
总的来说,用一种特殊的培养方法来模拟器官发育有缺陷的人类疾病,如无脑畸形,是可行的,这种方法结合了病人来源的iPSCs,模拟了器官发生的一部分。
换句话说,进一步发展人类ipsc衍生的三维类器官培养技术将极大地有助于理解人类的发展和疾病。