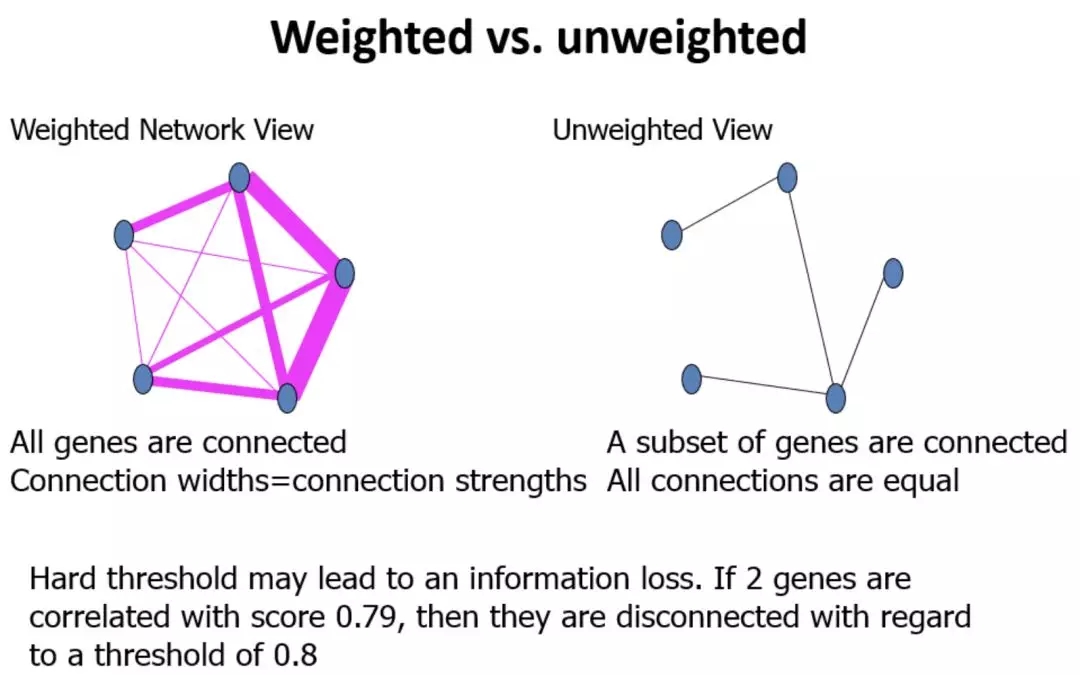
WGCNA 的输入矩阵到底是什么格式?
请问用 tcga 做 wgcna 分析,原始数据输入 tpm 和 fpkm 格式都行吗?
如果下的 raw_count 有 r 包转换吗?
首先,TCGA 目前的确是以 count 格式的矩阵下载为主
至于能不能找到 RPKM 这样的矩阵,肯定是可以的,但是我教大家的主要是 count 值,因为对 RNA-seq 数据的差异分析以这个 count 为 input。
然后问题就是,用 tcga 做 wgcna 分析,是不是原始数据输入一定要是 tpm 和 fpkm 格式?
(PS,类似的基因表达量的归一化还有很多,详细见:https://hbctraining.github.io/DGE_workshop/lessons/ 02_DGE_count_normalization.html)
那么问题就是,用 tcga 做 wgcna 分析,是不是原始数据输入一定要是 tpm 和 fpkm 格式?
其实呢,我最开始的教程,的确是 fpkm,所以大家会以为必须要这样的输入格式,详细教程见:一文看懂 WGCNA 分析 (2019 更新版)
实际上,WGCNA 首先会对全部基因的表达量计算两两之间的相关性,这个时候,只需要基因的表达量是适合计算相关性的即可,如果是 原始 counts 值,可以直接转为 log(cpm+ 1) 的格式 ,更为重要的其实是挑选多少个基因进入后续的 wgcna 流程。
以及我们的基因被 WGCNA 算法分成了不同模块后,哪些是有生物学意义的,跟表型相关性。
接着什么样的程序一定要 tpm 和 fpkm 格式呢?
类似 tpm 和 fpkm 的基因表达量的归一化还有很多,详细见:https://hbctraining.github.io/DGE_workshop/lessons/ 02_DGE_count_normalization.html 。
如果是需要对基因表达量进行排序,这个时候,基因长度就有影响,所以需要使用 tpm 和 fpkm,比如:http://xcell.ucsf.edu/。

最后如果下的 raw_count 有 r 包转换为 tpm 和 fpkm
其实我 GitHub 有代码的,而且我还提出了 3 种方法,全部代码如下:

上面的代码有点复杂,如果 R 语言水平不够,不建议去理解了。其它知识点代码是:https://github.com/jmzeng1314 /scRNA_smart_seq2。