小卫星可变重复序列 PCR,1 号染色体长臂(lq42〜43)的小卫星 MS32 即 D1S8 位点的等位基因内部含有碱基数目相同但序列不同的两种重复单位,其差别在于重复序列 3'端第二个碱基 G 突变为碱基 A,导致产生或消除 HaeⅢ的酶切位点,利用 PCR 技术分别扩增这两种重复单位,电泳分离并用数字编码其排列图谱,从而获得数字化的遗传信息,该方法被称作数字编码的小卫星可变重复序列 PCR 。MVR-PCR 具有 高灵敏度、高识别能力,尤其适合法医学中 DNA 多态性分析的需要。
小卫星可变重复序列 PCR 的基本原理是,以 Jeffreys 最初建立的 MS32 位点的 MVR-PCR 分析为例。MS32 位点由 29 个核苷酸的重复单位串联形成,重复单位有两种类型:a 型和 t 型,a 型含有 HaeⅢ酶切位点,t 型由于该酶切位点中的 G→A 的碱基突变而不能被 HaeⅢ切割。针对这些重复单位的特点,设计 4 种引物:①TAG-A,是自 HaeⅢ切点起与 a 型重复单位互补的寡核苷酸链,在 5』端又接上 1 段由 20 个核苷酸构成的不能与模板互补的 TAG 顺序,其作用是防止 MVR 的两种特异性引物在 PCR 产物内部引导扩增而产生进行性 PCR 产物的缩短;②TAG-T,它与引物 TAG-A 只相差 1 个碱基,是一段与 t 型重复单位互补的顺序,也接上同样的 TAG 顺序;③引物 TAG,它与 TAG-A 中的 TAG 顺序相同,只能与扩增后链中的 TAG 顺序退火(图 13-1); ④引物 32D 则选用小卫星 DNA 邻近的一段序列,其序列如下:5'-CGACTCGCAGATGGAGCAATGGCC-3',或选用与重复序列更加邻近的一段顺序 32O,其序列为 5'-GAGTAGTTTGGTGGGAAGGGTGGTT-3』。
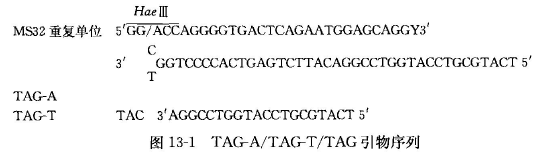
将样品分为 2 组:A 组和 T 组。加入 TAG 引物和 32D 引物后,分别加入 TAG-A 和 TAG-T 引物。TAG-A 和 TAG-T 作为下游引物单向扩增模板 DNA,获得带有 TAG 末端的扩增产物。32D 引物作为上游引物以带有 TAG 末端的扩增产物为模板进行 PCR 扩增产生带有 TAG 和 32D 末端的扩增产物。最后在 32D 和 TAG 引物作用下,扩增获得 A/T 型重复单位到 32D 区域的 PCR 产物群(图 13-2)。

当将 A、T 两组 PCR 产物经电泳分离后,将结果叠加,根据 Jeffreys 的编码原则,MVR 图谱中表现出 3 种重复单位:a 型在 A 行中出现带(在高加索人群中占 72.9%); t 型在 T 行中出现带(占 25.5%);。型在 A、T 行均无带。原因可能是重复单位内没有与 a 型或 t 型特异性引物相匹配的序列,这种概率很小,仅为 1.6%。样品基因组 DNA 的图谱是 A 行和 T 行谱带叠加的结果。根据叠加结果中带的强弱和有无分成 6 种等位基因,并分别用数字 1〜6 来表示。纯合子 中仅出现 1,2,6 三型。每个数字编码表示 1 个重复单元,含有相当丰富的信息量。
小卫星可变重复序列 PCR 的基本过程可分为如下几步:
(一)DNA 模板的制备
1. 样品来源
MVR-PCR 多应用于人类遗传学研究和法医学的 DNA 分析。其样品来源多样,一般用于人类遗传学分析的样品多为釆集的血样;而进行法医学检定的样品则为与犯罪有关的人体组织、体 液、分泌物、毛发、骨组织、精斑、唾液等物证。这些物证应依不同的种类和性质,按法医学标准程序提取、包装及保存。
2.DNA 提取方法的选择
选择何种基因组 DNA 提取方法应根据具体样品与 DNA 的后处理而定。最常用并且技术成 熟的 DNA 提取方法是酚-氯仿抽提法,该方法对样品用蛋白酶 K 进行消化处理,用酚-氯仿反复抽提获得 DNA;此外还有使用 NP-40、Triton X-100 的无酚提取法以及硅珠吸附法等。 不论釆用何种方法,均应简化操作步骤,缩短提取过程以减少 DNA 的降解,特别是毛发、精 斑、血痕等样品量很少而且核酸极易降解的法医学检材。
在对法医学检定的样品进行 DNA 制备时,应根据不同样品的各自特点进行一些必要的预处理,以提高 DNA 的产率;同时也应注意对 DNA 进行纯化,以避免这些预处理对 PCR 反应可能的抑制作用,例如骨骼样品的核酸提取,在进行脱钙预处理后,可能会残留影响 PCR 反应的钙离子,应将制备的 DNA 进行纯化,或者在提取过程中将钙离子去除。
DNA 提取也可直接使用现成的基因组 DNA 提取试剂盒,例如 QIAGEN 公司的 QIAamp®基因组 DNA 提取试剂盒系列,该系列针对从临床样品如血液及相关产品、毛发、精液及各种组织 中分离基因组 DNA。使用试剂盒获得的基因组 DNA 量与纯度更高,并且操作方便快捷。
下面介绍最常用的酚-氯仿抽提法,其它的基因组提取方法,如甲酰胺解聚法、异丙醇沉淀法、Chelex100 抽提法、NP40 裂解法、Triton X-100 裂解抽提法、硅珠抽提法,可参看相应文献;各种基因组 DNA 提取试剂盒使用方法可参看相应试剂盒手册。
3. 酚氯仿抽提法
(1) 样品的处理
①组织:取 1〜20 mg 动物组织,去除结缔组织和残血后,移入预冷的研钵中,用力快速研 磨成匀浆。如果是富含 DNA 酶的胰脏、脾脏,或者富含胶原蛋白的皮肤、肌腱等组织,以及坚 硬的骨骼等,应在研磨中反复添加液氮,直至将组织研磨成粉末状。加入 600 风细胞裂解缓冲液 [10 mmol/L Tris-HCl (pH8. 0), 100 mmol/L NaCl, Immol/L EDTA (pH8. 0)], 继续研磨 lmin, 将组织匀浆转移到 1. 5 ml 离心管中。
注意:进行液氮操作时,需小心操作防止冻伤。如果处理的样品为有害生物材料,例如含有致病菌等, 则应按相应等级生物安全操作规程进行,并对器械和废弃物进行处理。
②细胞:收集 5×106 个数目的细胞,用 PBS 磷酸盐缓冲液或者生理盐水漂洗 2 次。用 600μl 细胞裂解缓冲液悬浮。
③血液:将 1 mL 新鲜抗凝血,1300 g 离心 15 min, 弃上清 (血浆),将淡黄色下层小心移至新的离心管中,加双蒸水洗细胞 2 次,弃上清。如果是冻存血液,将血液室温解冻后,加入等体积 PBS 混匀,3500 g 离心 15 min, 弃上清。用 600μl 细胞裂解缓冲液悬浮白细胞。
注意:全血中唯一含有细胞核可用于制备基因组 DNA 的血细胞为白细胞,仅占总量的 0.3%。一般正 常人的白细胞数目为 107 个/mL 血液,但有些病人血中白细胞数目可能是这个的 10 倍,因此如果使用试剂 盒从病人全血中制备基因组 DNA 时,需要减少血液样品的体积以免超过提取体系的承受范围。
此外采血液标本时,应注意从静脉血与动脉血提取的 DNA 的量会有所不同。
(2)DNA 的提取
在 600μl 体系中,加入 60μl SDS (100 g/L) 和 10μl 蛋白酶 K (10 μg/mL)。37°C 过夜或者 56°C 保温 4 h (保温过程中,应不时轻轻摇匀反应液) 裂解细胞,消化蛋白。将反应液冷却至 4°C, 加等体积 Tris 饱和酚 (pH8.0), 充分混匀,4°C10000 g 离心 10 min, 吸取上层黏稠水相移至另一离心管。加等体积 Tris 饱和酚 (pH8.0),重复一次 Tris 饱和酚抽提。吸取上层水相,加等体积氯仿-异戊醇 (24:1, 体积比),充分混匀,4°C10000 g 离心 5 min。吸取上层溶液移至另一离心管。重复一次氯仿-异戊醇抽提。吸取上层溶液至 2.5 倍体积冷 95% 乙醇中,轻摇至 DNA 析出。离心弃上清,用 70% 冷乙醇清洗 3〜4 次。加适量 TE 溶液 [10 mmol/L Tris-HCl, lmmol/L EDTA (pH8. 0)] 或者去离子水溶解 DNA。
(二)MVR-PCR
1.MVR 位点与相应引物设计
目前已发现的 MVR 位点有 D1S8(MS32),D7S21(MS31A), D16S309(MS2O5),MSY1(DYF155Sl)g 等。设计 MVR-PCR 引物时,应根据研究目的和方法的不同设计相应的引 物(表 13-1)。
表 13-1 已应用的 MVR-PCR 引物
| MVR 位点 |
引物 |
| D1S8(MS32) |
32D: 5'-CGACTCGCAGATGGAGCAATGGCC-3』
32-TAG-A/T:3'tcatgcgtccatggtccggaCATTCTGAGTCACCCCTGGCC/T) -5'
TAG: 3'-tcatgcgtccatggtccgga -5'
|
| D7S21(MS31A) |
31-Alu 1+/-, 31 -Psp14061 + /—, Hga I +/-
31-TAG-A/G, TAG
31A; 5'-CCCTTTGCACGCTGGACGGTGGCG-3'
31A-A/T: 5'-AGTGTCTGTGGGAGGTGG(A/G)-3'
|
| MSY1(DYF1S5S1) |
Y1A: 5'-ACAGAGGTAGATGCTGAAGCGGTATAGC -3'
TAG1/3: 5'-tcatgcgtccatggtccggaTGTGTATAATATACAT C/G ATGTATATTG -3'
Y1B: 5'-GCAACTCAAGCTAGGACAAAGGGAAAGG -3'
TAG3R/4R: 5' - tcatgcgtccatggtccggaCATCATGTATATTATACA C/T AATATACATC-3'
|
早期的引物设计中,除在重复单位上游侧翼序列中设计上游引物(如 32D, Y1A 等)以及 分别扩增不同类型重复序列的下游引物(如 TAG-A/T 等)外,还在扩增反应中使用 TAG 引物。 现在基本省略 TAG 引物,直接使用上游引物分别与 A/T 引物扩增,同样可获得理想的不同类型重复单位的扩增产物图谱。
2.PCR 反应
根据重复单位的类型和研究需要,每个标本设置不同的反应管,每管反应体系可设为 50μl, 加入 50 〜100ng 基因组 DNA, 2 .0mol/L MgCl2, 5μl 10 X PCR 缓冲液 [500 mmol/L KC1, 200 mmol/L Tris-HCl(pH8.3)] 0.2 mmol/L dNTP, 56. 5 μg/ml BSA, 20μmol/L 上游引物(如 32D, 31A, Y1A 等)(或者 20μmol/L 上游引物与 20μmol/L TAG 引物),以及 Taq 酶 1.5U。分别在不同的管内加入 20μmol/L 相应的下游引物。加水补至 50μl 总体积。如果所用 PCR 仪无加 热盖,则须在反应管中滴加石蜡油覆盖。
在 PCR 扩增仪上按下列条件反应:先 94°C 预变性 4 min,然后 94°C 变性 1 min, 68°C 退火 1 min,72°C 延伸 5 min,共 25 个循环,最后 72°C 延伸 10 min。
引物和所扩增产物不同,PCR 反应条件也有所不同。应根据具体的扩增需要,对反应条件 进行相应调整和优化。
3. 电泳
PCR 产物在 35 cm 长的 1% 琼脂糖凝胶,0.5%TBE 电泳缓冲液 [45 mmol/L Tris-硼酸, 1 mmol/L EDTA] 下以较低的电压进行电泳分离。
4. 探针标记、杂交与检测
电泳分离后,将凝胶取出,并切除边缘无用的部分。在凝胶左下角或右下角切去一小三角形以作为操作过程中凝胶的方位标记。将凝胶中的 DNA 经碱变性后,真空转移到尼龙膜上(或其它转移方法)。釆用中性缓冲液转移获得含有靶 DNA 的尼龙膜需要采用真空烘烤、微波加热或者紫外照射等方法将 DNA 固定在尼龙膜上。采用碱性缓冲液转移则不需要 DNA 的固定。
选择小卫星重复序列共有的碱基序列作为杂交探针。将含有靶 DNA 的膜与 32P 标记的寡核苷酸探针杂交。65C 反应 3 h。室温 X 射线片放射自显影 6 h, 即可得到 MVR 图谱。
如果在 PCR 反应液中添加放射性标记的核苷酸(如 [α-32P] dCTP), 可将 PCR 产物经琼脂糖凝胶电泳或者中性聚丙烯酰胺凝胶电泳分离。可直接用湿胶或者干燥的凝胶进行 X 射线片压片放射自显影 8〜12 h(凝胶干燥后放射自显影灵敏度会有少许提高),得到 MVR 图谱。
也可采用辣根过氧化物酶标记的寡核苷酸作为探针,以避免放射性污染。步骤如下:取探针配成 12ng/μl 使用液,加入等体积的辣根过氧化物酶标记试剂,混匀后加入等体积的戊二醇,混匀,快速离心。37°C 水浴处理 30 min 后与含靶 DNA 的尼龙膜 37°C 杂交 1 h。杂交后的膜在洗膜液 [6mol/L 尿素,0.5XSSC, 0.4%SDS] 室温振荡洗膜 2 次,每次 10 mino 然后在 2XSSC 溶液中室温洗膜 2 次,每次 5 min。最后进行 X 射线片曝光,得到 MVR 图谱。
具体的标记、杂交以及放射自显影等操作细节可以参看《分子克隆实验指南》(第三版)第六章及附录 9 检测系统等相关章节他。
(三)MVR 图谱的数字编码
将获得的 MVR 图谱上的 A 与 T 的结果叠加,根据 MVR 数字编码原则进行编码,即可得到 特异的 MVR 编码。
MVR-PCR 的 6 级数字编码原则如表 13-2 所示。
表 13-2 MVR-PCR 的 6 级数字编码
| 编码 |
MVR PCR 电泳谱带特征 |
|
| |
A 行 |
T 行 |
| l(aa) |
强 |
无 |
| 2(tt) |
无 |
强 |
| 3(at) |
弱 |
弱 |
| 4(a0) |
弱 |
无 |
| 5(t0) |
无 |
弱 |
| 6(00) |
无 |
无 |
如果忽略弱带与强带的差别将编码 4 与 5 视为 1 与 2, 得到 4 级编码(表 13-3)。
表 13-3 MVR-PCR 的 4 级编码
| 编码 |
MVR-PCR 电泳谱带特征 |
|
| |
A 行 |
T 行 |
| 1(10) |
有 |
无 |
| 2(01) |
无 |
有 |
| 3(11) |
有 |
有 |
| 6(00) |
无 |
无 |
该编码方法不考虑谱带的强弱变化,虽然减少了 MVR 分析的信息量,但是避免了判读的主 观性,并且经过实验室和实际应用验证,该编码方法获得的数字编码对 MVR 个体识别能力没有 大的影响,足以满足法医学的一般的 DNA 分析所需。







