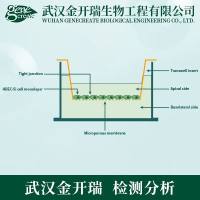
材料与仪器
步骤
一、材料
1. 胞质分裂阻滞微核试验

2. 微核的着丝点检测
(1)从硬皮病 C R E S T 亚型的患者中取得的血清样本。
(2) F I T C 标记的兔抗人 I g G 二抗。
(3) 过氧化物酶标记的兔抗人 I g G 。
(4) 二胺基联苯溶液(D A B ): I m g /m L 溶于 0. 5 mol/L Tris 碱缓冲溶液, p H 7. 6。
(5) NiCl2 溶 液 : 8% 溶液溶于 0.5 mol/L Tris 碱缓冲溶液, p H 7. 6 , 使用前配制。
(6)D A B 反应混合物: I m L D A B 溶 液 , 3 m L Tris 碱缓冲 溶 液 , p H 7.6, 25NiCl2 溶液 , 40 uL 0 •I m o l / L 咪唑水溶液和 10 u L 30% 过氧化氢。使用前配制。
(7)中性红溶液: 0.1 % 溶于双蒸水。
二、方 法
1. 用于分离的人淋巴细胞的标准 CBMN 试验
在这个实验中只有出现在用 P H A 刺激后完成一次核分裂的细胞中的 M N i 才用于分析。这些细胞可通过由在第一次有丝分裂之前加入 Cyt B 使细胞不再进行后续细胞分裂而导致的双核形态来辨认。在优化条件下 P H A 刺 激 后 72 h 在 所 有 活 细 胞 中(除去坏死和凋亡外的所有细胞)双核细胞比例可达 3 5 % 〜6 0 % 或更多。所有实验设备都必须
达到生物安全要求以保护操作者,并且所有在此程序中使用的溶液需经过滤灭菌。
1.1 淋巴细胞的分离、细胞培养和细胞收集
(1)用含有抗凝剂肝素的管子收集新鲜静脉血,置 于 22°C , 4 h 内分离。
(2)用已灭菌的 0. 8 5 % NaCl 按 I : 1 稀释血液,轻轻颠倒混匀。
(3)将稀释后的血液轻轻覆盖在 Ficoll Paque (Pharmacia) 密度梯度上,所用比例大 约 1 : 3 (例 如 2 mL Rcoll Paque 加 人 6 m L 稀释的血液),小心不要扰动两相界面。
(4)在 22°C 下 400 g■离心 25〜40 min。
(5)淋巴细胞位于 Ficoll Paque 和稀释的血浆之间。收集后在 22°C 加 人 3〜5 倍体积的 HBSS。根 据 体 积 的 不 同 情 况 ,将 所 得 细 胞 悬 液 在 280〜 400 g 下离心5〜IOmin0
(6)弃上清,用 2 〜5 倍 体 积 HBSS 重 悬 细 胞 ,并 根 据 不 同 体 积 用 180〜400 g 离心 5 min。
(7) 弃上清,用 I mL RPMI1640 培养液重悬细胞。
(8)细胞计数计算细胞浓度,根据台盼蓝实验得到的活细胞比例调整细胞浓度。
(9) 用 含 有 1 0 % 〜15% 灭 活 胎 牛 血 清 的 RPMI1640 培 养 液 调 整 细 胞 浓 度 至 0. 5 XIO6〜I .OXlO6 细胞/mL, 并 在 圆 底 的 培 养 管 中(10 m m 宽)用 0.75〜1.0 mL培养液进行培养。
(10)每个培养管中加入 10 ML/mL PHA (Glaxo Wellcome HA1 5 ) 刺激淋巴细胞分裂。将盖拧松, 37°C , 5 % C0 2, 湿润条件下培养。 P H A 的浓度需根据产品的纯度和来源进行优化,以 保 证 细 胞 在 用 Cyt B 阻滞后产生最大数量的双核细胞。
(11) P H A 刺 激 44 h 后 ,每毫升培养液加人 4.5 pg CytB (戴手套和通风橱内操作):溶 解 IOOmL Cyt B 储 液 ,加 入 900 培养液,混合。将 75 混 合 液 加 人 1m L ±普养液使终浓度为 4. 5 Mg Cyt B/mL (—些实验室也成功地使用了 6. 0ugC y t B /m L 的浓度)。继续培养。
(12)加入 C y t B 28 h 后用血细胞分离器(cytocentrifuge, ShandonElliot) 收集细胞。弃去 1 〇〇 uL 培养液,将细胞轻轻重悬于管中。将 100 〜120ul 细胞悬液转移至血细胞离心杯(ShandonElliot) , 离心至每玻片产生两个小点(见注释1)。(如下设置细胞离心程序,时间: 5 min,转速: I O O fiO 将玻片从血细胞分离器中移开,空气干燥 1 〇 〜2 〇 m i n, 用 1 0 0 % 甲醇固定 l 0m in 。
(13)用不同的可区分细胞核与胞质界限的方法染色。在我们的实验中使用的是「DiffQuik」,它是一种商业成品,能迅速、优化地给出结果(见注释 1)。
(14) 染色后将玻片晾干并用 D P X 液盖上盖玻片。这一步骤需在通风橱内完成,将片子放在通风橱内储存,直到需要使用。
对照和经遗传毒物处理的细胞培养需有重复样本 ,每个样本都制备玻片。这对实验数据标准差的获取来说是必需的,例如变异系数(标准差),对于每组重复数据都需要标明( 见注释 2 和 3)。这一实验设计见图 2。
对于突光显微镜,推荐使用吖陡橙(40 ug/mL溶 于 Sorensen』 s 憐酸缓冲液, p H 6 . 9 ) 进行染色。如果没有细胞离心机,可使用下面介绍的用于全血培养的方法准备玻片

1.2 玻 片 的 检 查 以 及 M N 频率的评价
最好用放大 1000 倍的光学显微镜或荧光显微镜观 疲 诚 士 诚 屮 單 女 公 观察玻片。玻 片 最 好 在 分 析 _代 号 标 记 ,这 样 分 析人员不会知道玻片的含义(盲法)。分析数据需两位不同的分析者用相同显微镜对每个重复实验进 行 评 判( 见注释 4 和 5)。进行统计的细胞数( 见注 释 6 ) 需根据实验预期检测 M N 指数的变化值和预期的标准差确定。每张玻片需获得
以下信息:
(1)计算至少 1000 个 双 核( B N ) 细 胞 内 的 微 核(M N i ) 数量以及 M N i 在 1000 个B N 细胞内的频率。在 B N 细胞中计数 M N i 的标准如下。
(2)含有 0、 1 或更多微核(M N i ) 的 B N 细胞的分布;一个双核细胞中的 M N i 数在正常人体淋巴细胞中约为0〜3 个 ,但根据毒物暴露和年龄的不同有时可能大于3 个。
(3)在至少 1000 个 B N 细胞中出现微核的 B N 细胞频率。
(4)统 计 1000 个 B N 细胞中细胞核质桥的频率。在 B N 细胞中计数核质桥的标准如下。
(5) 每 500 个细胞中单核、双核、三核和四核细胞的比例。从此信息可以得出核分裂 指 数(nuclear division index,稍后解释)。
(6)在统计活的单核、双核或多核细胞频率的同时,统计在同张玻片上每 500 个细胞中由凋亡或坏死引起的已死或将死的细胞比例( 评判标准稍后列出)( 见注释7)
需要注意的是当无法判断如何对某一细胞进行分类时最好忽略该细胞。一张统计表中应包含的基本信息见表 1。

1.3 分 选 双 核 细 胞 的 标 准 及 微 核 、细胞核质桥、核芽、凋亡和坏死的细胞的计数
(1)双核细胞的分选标准:用 于 M N 频率计算的胞质分裂阻滞的细胞须符合以下特征:
a. 细胞须为双核。



b. 在一个双核细胞的两个细胞核须有完整的核膜,并须在一个细胞质内。
c. 双核细胞中的两个核的大小、染色形状和密度应相似。
d. 在 B N 细胞中的两个核可能由不超过 1/4 核直径的细胞核质桥连接。
e. 在 B N 细胞中的两个核可能相互接触,但是不该重叠。只有在这两个核的界限非常清晰时才能将此种重叠的双核细胞计算在内。
f. 双核细胞的胞质界限或细胞膜必须完整,并能与附近细胞清晰区分。
可以或不可以用于计数的双核细胞的类型如图 3 所示。 在计算微核细胞频率时不应被包括的细胞种类包括单核、三核、四核和多核细胞,以及凋亡或坏死细胞( 图 4)。
-
微核计数的标准: M N i 在形态上和核相似,只是比核小。它们还有以下一些
特点:
a. 人淋巴细胞 M N i 直径的大小通常占双核细胞中主核直径的 1/16〜1/3, 其面积相当于主核的 1/256〜1/9。
1.4 核 分 离 指 数 计 算( N D I ) 以 及 核 分 离 细 胞 毒 性 指 数( NDCI)
(1)根 据 Eastmond 和 Tucker 的 方 法(3 0 ) 计 算 N D I 。计 数 500 个活细胞,计算含有一个、两个、三个或四个核的细胞频率, N D I 计算公式如下:
N D I = ( M 1 + 2 X M 2 + 3 X M 3 + 4 X M 4)/N
M l 〜M 4 指含有 1〜4 个细胞核的细胞, N 表示总活细胞数。 N D I 指数以及双核细胞的比例是比较淋巴细胞的有丝分裂反应以及测试药物的细胞抑制作用的有效参数。
(2)用于评价核分裂状态的更为准确的方法是将坏死和凋亡的细胞数包含在细胞总数内,因为在高剂量的毒性物质作用下死细胞可能占了很大的比例。所以需要注意的是如果不包括坏死和凋亡的细胞,双核细胞比例和 N D I 指数会偏高。
(3) 下面经修正的公式可用于较为准确地估算核分裂状态以及细胞分裂动力学的情况 ,这个公式考虑了坏死和凋亡的细胞。
NDCI= (A P + Nec+ M l+ 2X M2+ 3 X M3+ 4 X M4) /undefined
NDCI: 核分裂细胞毒性指数(nuclear division cytotoxicity index); A p : 调亡的细胞; N e c : 坏死的细胞数; M l 〜M 4 指含有 I〜4 个细胞核的细胞; N *表示总 细 胞 数( 活细胞和死细胞)。
2 用阿糖胞苷微核实验检测人淋巴细胞 (V G 1 期 DNA 剪切修复损伤的方法
在对暴露于各种遗传毒物的人 G 。期淋巴细胞的 M N 进行检测后发现,对于主要引起碱基损伤和 DNA 加合物而不是 DNA 链断裂或者纺锤体损伤的化学物质和紫外辐射而言, MN 的形成与细胞毒性相比明显程度较低(2 1 ) 。 我们猜测这可能是由于对损伤的有效修复,或者未修复的损伤位点在一轮 DNA 合成后不会形成 DNA 双键断裂。因此 ,我们推测用阿糖胞苷(ARA) 抑制剪切修复会使碱基损伤转化为单链断裂继而在DNA 合成后变成双键断裂,导致无着丝点片段的产生并将在一个分裂周期内以 MN 的形 式 出 现(21, 31)。
(1)根据这一概念( 图 8),我们在淋巴细胞培养的第一个 16 h 内( D N A 合成前)加人 A R A (1 ug/m L 培养液)的确可增高紫外照射或 M N U 处理 后 M N 的形成(10 倍或更高)。而在 X 射线暴露后加人 A R A 引起的 M N 数量变化仅为 1. 8 倍 ,因为 X 射线引起的 D N A 加合物或碱基损伤与 D N A 链的断裂相比较少。这种方法已用于判定可引起剪切修复的杀虫剂,来区分诱导或者不诱导剪切修复的遗传毒物。

(2) A R A 实验是对 C B M N 实验重要的补充,特别是当观察到强烈的细胞毒性作用和较弱的 M N 产生时更需要做 A R A 。
(3)只有经过 C B M N 实验才能用 A R A 方法准确检测 D N A 剪切修复损伤,这是因为(a) 只有在核分裂完成的情况下才发生 D N A 剪切修复损伤向 M N 转变;(b) A R A 的加入可能引起细胞分裂动力学的改变,若没有 Cyt B 则可能在 M N实验中产生干扰结果。
(4) A R A 抑 制 D N A 聚合酶,因此可能导致细胞在进行 D N A 复制合成时产生 D N A双链断裂。因此,这种方法仅在 P H A 刺激的 G 。期淋巴细胞,在 G 1 期、 S 期前暴露于 A R A 才可以,因为剪切修复通常在 G 1 期被激活。
(5) 实际操作中这意味着细胞在 P H A 刺激后的 16〜20 h 内加入 A R A ,之后洗细胞去除 A R A ,在含有脱氧胞嘧啶的培养液中培养,逆 转 A R A 对 D N A 聚合酶的抑制。
(6)以上步骤后进行标准的 C B M N (如前所述)。更详细的流程和典型结果可在 Fenech 和 Nexille ( 2 1 ) 及 Surrales 等(3 2 ) 的文章中找到。
3 其他细胞培养体系的 CBMN 实验
3.1 淋 巴 细 胞 的 全 血 细 胞 培 养
(1)用全血培养也可以进行人淋巴细胞的 C B M N 实验。
(2) —般 0 •4〜0 •5 m L 的全血加人 4. 5 m L 的 培 养 液(如 R P M I 1640),含有胎牛血清、谷氨酸、抗生素和 P H A 。
(3)在 P H A 刺激后 44 h 加人 Cyt B 。全血细胞培养中推荐的 Cyt B 最优浓度为 6 u g/m L (33)。
(4)加入 Cyt B 后 28 h 收集双核淋巴细胞,过程如下:
a. 300 g离心 5 min 收集细胞,弃上清。
b. 用 7ml 4℃预冷的 0.075mol/L KCI低渗处理以裂解红细胞, 300 g离心 8m i n
C . 弃 上 清 ,加 人 5 m L 甲醇- 乙 酸( 3 : 1 ) 固 定 液( 必 须 一 边 摇 晃 一 边 加 人 固 定液 以 防 止 块 状 物 形 成 。)
d. 然后以 300 g 离心 8 min, 用固定液继续洗两遍。
e. 轻轻重悬细胞,将重悬液滴在玻片上晾干。
此外也可以用 Ficoll 梯度液从全血细胞培养中直接分离双核淋巴细胞,然后在固定和染色前通过离心转移细胞至玻片,这种方法会避免低渗处理,以更好保留细胞质。
(5) 用内含 1 0 % 吉姆萨染液的 0.1 m 〇 l/L 磷酸钾缓冲溶液(p H I 3 ) 染色,用于光镜下使用。或者用吖啶橙(含 l O ug/m L 的 0. l m o l /L 憐酸缓冲溶液, pH 6. 9,用于荧光显微镜下使用)。
3.2 鼠 淋 巴 细 胞 培 养
(1)从脾脏或外周血中分离淋巴细胞,根据 Fenech 等的描述培养(34)。
(2)因为鼠的淋巴细胞比人淋巴细胞的分裂周期短,因此需要在分裂素刺激后 i8 h内加入 C y t B , 并在 20 h 后收集细胞。根据培养条件的不同,甚至可以在分裂素刺激后 72 h 后得到较好的双核细胞比例
3.3 其他原代细胞培养包括肿瘤细胞培养
CBMN 实验可用于其他细胞类型来检测 DNA 损伤,包括体外、体内或者来自体内的体外实验。最重要的是记住以下几点:(1)确保计数的 M N i 是在遗传毒物作用后的第一次核分裂中产生的;(2)做预实验以确定 Cyt B 的浓度和孵育时间,以获得最多的阻滞在双核期的分裂细胞(见注释 10)。此外需要记住的是 Cyt B 可能需要 6 h 才能发挥阻滞胞质分裂的作用(未发表的数据)。
a.用建立的细胞系或者从分裂细胞群中得到的原代培养细胞系时,通常在遗传毒物作用后立即加入 Cyt B 以得到所有进行第一次核分裂的双核细胞—收集细胞前通常需要 24〜48 h 的孵育时间,取决于细胞周期的时间。
b.贴壁细胞先要消化,再经如前处理人淋巴细胞所述的血细胞分离离心。具体方法已在核骨髓细胞(14)、肺 纤 维 细 胞(1 5 ) , 皮肤角蛋白细胞(3 6 ) 和原代肿瘤细胞培养(1 3 ) 中有介绍。
c.为了分析体内诱导的 MN, 更具操作性的方法是在将细胞从动物体内分离后置于含有 Cyt B 的细胞培养液中来阻滞分裂细胞的胞质分裂。这种方法已在多种细胞包括纤维细胞、角蛋白细胞、有核骨髓细胞等中得到成功应用。
4 胞质分裂阻滞或不阻滞的细胞系或培养肿瘤细胞中的微核实验
(1)关于用于积聚双核细胞的 Cyt B 是否会影响 M N 的表达还有争议(28)。正常细胞的研究没有发现 Cyt B 可诱 导 M N i ; 在通常用于阻滞胞质分裂的浓度下, CytB 也未显示有引起 M N 出 现 频 率 增 多 的 剂 量 效 应 (1 〇 , 37-39)。最近研究表明在胞质分裂阻滞的 B N 细胞中由纺锤丝毒物引起的 M N 表达可能比预期的少。因为两极之间缩短的距离会增加滞后的染色体片段或者整个染色体回到核内的几率。但 是 ,这并不影响 C B M N 实 验 的 有 效 性 (40)。
(2)越来越多的人将眼光投入到开发不使用 Cyt B 的体外 M N 实验中,以减小 Cyt B的影响,但是由于缺乏细胞分裂动力学对照很可能产生假阴性(如抑制核分裂会 妨 碍 M N 的表达)。
(3)虽然缺少普通细胞 CBMN 实验可造成假阳性结果的证据,但已有很多证据表明不考虑核分裂阻滞的 M N 实验会引起假阴性结果,或者低估人淋巴细胞中的MN (10, 11, 41),不 用 Cyt B 的 M N 实验的缺陷在图 9 中举例说明。
(4) 尽管如此,近期研究比较了用 Cyt B 或 不 用 Cyt B 的 M N 实验后,认为如果细胞生长状态很好且培养和核分裂合适的情况下,在检测强断裂剂时,无 Cyt B的条件下可得到相似的 CBMN 和 M N 结 果 (42, 43)。
(5) M N 表达的数学模型预测 (1 ) 在双核细胞中 M N 计数是计算 M N 频率最可靠的方 式 (2 ) 当核分裂被测试的化学试剂显著阻滞或培养条件下无法得到理想的分裂细胞时,在 不 用 胞 质 分 裂 阻 滞 的 单 核 细 胞 中 M N 计 数 会 引 起 假 阴 性 结 果(44)
(6)因此,用不经 Cyt B 处理的单核细胞得到的 M N 频率不能用作实验的结论性结果 ,需通过 CBMN 实验的阴性结果来进一步证实。

5 测量微核和不分裂中的染色体丢失的分子技术
要充分利用 C B M N 实验的优势就需要区分来源于整个染色体的 M N i ,以及来源于无着丝点片段的 M N i 。这能通过针对着丝点 D N A 的特异性探针或者针对聚集在活跃的染色体着丝点区域的着丝点蛋白的特异性抗体来实现。对于人类细胞或其他染色体大小为异质的细胞用 M N 的大小来做判断并不合适,因为小 M N 可能包含大染色体的片段,或者一整个小染色体。最简单且经济的方法是用抗着丝点抗体 (45),但这种方法不能区分特定染色体,并且可能无法探测由不活跃中心粒的着丝点蛋白缺失而发生的染色体丢 失 (46)。用 原 位 杂 交(I S H ) 确定中心粒区域虽然费钱费力但它有更好的特异性;例如 ,可以利用针对特定染色体的着丝点探针探测在双核细胞中的不分裂事件(在子核中同源染色体的不平等分离)(17)。本章只介绍着丝点蛋白抗体法。对 于 用 1S H 探测中 心 粒 区 域 请 参 考 Farooqi ( 1 7 ) , 以 及 H ando ( 1 8 ) 、 Ehajouji ( 2 3 、 4 7 ) 、 Schuler
5.1 玻片准备
( 2 5 ) 等的文献。用不同的技术预期可检测到的各种结果如图 1 〇所本。
1 CBMN 实 验 中 M N i 的 着 丝 点 检 测(1) 用同标准 C B M N 实验方法收集 B N 细胞,用血细胞离心器转移至玻片,空气干燥 5m i n , 用甲醛固定 IOmin, 再次空气干燥。
(2) 在此步骤,玻片可以立即使用或者放在密封的干燥盒子里储藏在液氮的气相部分 ,最长储存期限为 3 个月。
(3)要检测储藏的玻片上的着丝点蛋白时将玻片从液氮中移出,放在密封的盒内室温下平衡。
5.1.2 着丝点蛋白检测
(1)抗着丝点蛋白血清可以商业购买或者从有硬皮病 C R E S T 亚型病人血清的免疫诊所 获 得 (48)。用后者血清需要获得人类伦理方面的认可并且得到捐献病人的同意。
(2) 血清需要在培养细胞的中期分裂细胞涂片上,用 F I T C 标记的兔抗人二抗在荧光显微镜下观察检测。只有特异地与中期染色体着丝点反应的血清才能用于后期实验。(3)用 F I T C 标记的二抗检测着丝点蛋白虽然较为直接但需要荧光显微镜,并且为非永久制备的玻片。这一突光技术在它处已有详细描述 (45)。另一种可选方法是用免疫过氧化物酶染色,这可获得永久保存的玻片 (49),这种方法作为常规检测更实用,将在下一段中详细描述。
(4)在荧光过氧化物方法中,将固定好的玻片置于装有用 Tris& 缓 冲 液(P H 7. 6,6. O g Tris 碱/L 盐溶液)按 1 : 4 0 比例稀释的抗着丝点蛋白一抗的潮湿小室内,20℃孵育过夜。
(5)用稀释的正常人血清制备阴性对照。
(6) 第二天用同样的稀释抗体的 Tris 盐缓冲液浸没玻片 30 s 以冲洗玻片
(7)不用干燥,浙干玻片,用过氧物酶标记的兔抗人 I g G 抗体孵育 3 h 。
(8)再次沥干玻片,准备过氧物酶组化反应。
(9)免疫组化法中可给出最好对比的是能产生黑色沉淀的咪唑氯化镍改进的标准D A B 反 应 (50, 51)。
(10)在使用前配制 D A B 反应物,然后用 0.22 的滤纸过滤,以防止非特异的沉淀物出现在玻片上。
(11)玻片需要成批染色,包括阴性对照。
(12)在 20°C 反 应 Imin, 然后停止染色,将玻片放在水里漂洗。
(13)然后风干玻片,用核染料中性红 (0 . 1 % 水溶液)染 色 约 30s, 用水漂洗,风干,以制备永久玻片。
5.1.3 计 数 流 程
(1)只限于在每个核内至少含有 20 个着丝点的双核细胞中计数 M N i 着丝点的情况。
(2)按照是否含有着丝点将至少 100 个 M N i 进行分类,并且要注明每个 M N 含有的着丝点数。
(3)用以下公式计算含着丝点的 M N i 的最终比值:(朽一 P c ) / (l—P c ), P c 指玻片上正常血清处理后过氧化物酶反应为阳性的 M N i 的比例, P s 指玻片上抗着丝点蛋白血清处理后过氧化物酶反应为阳性的 M N i 的比例。
6 体外化学敏感性测试的处理流程
(1)理想状态下每一种化学物质都需在细胞周期的各种时期测试其潜在的遗传毒性。因为在收集时人外周血淋巴细胞都处于 G fl 期,对于评价此阶段的损伤而言较为理想。
(2) 然而,由于在 S 期 、 G 2 期和 M 期的细胞对于遗传毒性的作用更敏感,因此最好在大多数细胞分裂时将它们暴露于毒物中。因为 M N 表达需要一次核分裂的完成,所以在处理细胞和收集细胞之间需要有充足时间保证分裂的完成。
(3) G 。期处理的人外周血细胞需要尽早收集双核细胞,并且在尽量长的时间内收集以确保甚至是减数分裂延迟的细胞也能被观察到。经典做法是在 44 h 后加入 CytB ,在 72 h 后收集细胞,间隔的时间应该足够满足上述条件。但是也可用下述方法使分裂较晚的细胞能被观察到: 24 h 后加人 C y t B , 96 h 后收集细胞。
(4) 如果需要在 S 、 & 和 M 期进行处理,比如肿瘤细胞,需在细胞呈指数增长时加入化学物质,之后较短时间内加入 Cyt B 积累分裂细胞,再根据要检测的细胞周期在 6〜24 h 后收集细胞。
(5)早期收集的双核细胞多是处于 G 2 期或者 S 期暴露的细胞,而在晚些时候收集的双核细胞包括在各期暴露的细胞。因此相对于 Cyt B 加入时间而言,收集细胞的时间会影响观察到的细胞类型。
(6) 表 2 是 C B M N 实验检测遗传毒性的常规步骤。
(7)在测试新化学物质时可选择像 S9 混合物这样的代谢激活系统。但这可能会缩短暴露的时间,因为 S9 对靶细胞可能有细胞毒性作用。较好的方法是选择有代谢活性的细胞,例如基因修饰的 M C L -5 细 胞 (52)。

注意事项
(1)在我们进行 CBMN 实验过程中,最容易产生问题的是在玻片的制备和染色环节 ,因为计数结果的好坏依赖于制片的质量。主要注意事项:(a) 在将细胞置于玻片前应轻轻吹打,避免细胞结块;
(b)细胞密度适中,这样才容易辨认细胞质边界;
(c) 在染色所有玻片前先试染一张,以确定染色合适。
(2)要做重复以确保结果可信,同时也可计算组内重复标准差数据。细胞遗传毒性实验需要满足严格的数据分析标准,如 果 C V 值大 于 1 0 % 就应剔出。由于是肉眼计数观察实验,较大的标准差是可以接受的。根据我们的经验和国际实验室间计数比较的结果,基线数据 C V s 大于 4 0 % ,则不能接受;对于暴露于放射条件下培养,每 1000 个 BN 细胞中有大于 100 个 M N 时, C V s 应小于 2 0 % 。
(3)不熟练操作者(如学生和新技术员)的计数不可靠,除非他们在计数标准对照玻片时 C V 值达到可被接受的程度(不大于 40% ) 。
(4)不同计数人员间的差异是造成 M N 实验差异的主要原因之一 (71)。此,在一项研究中使用同一个技术员是很有必要的,最好有两名技术员,用如 图 2 所示的同样方法对重复组进行计数。另一方法是用有「低」、「中」、「高」 M N 频率的标准片对计数员进行校正。每一个计数员对标准玻片的计数可被用来计算一个校正值。这种方法虽然仍在完善中,但由于它考虑了在同一个实验室或实验室之 间计数员的视觉差异,从而也是值得重视的。
(5) 另一造成计数员和实验室间差异的重要影响因素是显微镜的质量和镜头。根据我们的经验,统计核质桥受到显微镜的影响,因为细小的核质桥会被质量较差的镜头忽 略。需要注意的是计数员应避免在实验中变换显微镜,并且实验室负责人应统一显微镜的镜头,并将其升级至一个较高的水平。我们推荐使用可以提供优质清晰度、 图像对比度和平整度的系统。系统应有放大 1000 倍的能力。
(6)常见问题之一是确定在 CBMN 实验中 BN 细胞的数量。虽然记数从 500 到 2000个 BN 细胞都有过报道,被认可的做法是每次处理或时间点计数不少于 1000 个BN 细胞。另一种方法是计算 B N 细胞数直到出现固定数量的微核(例 如 4 5 个微核)。后者的优点是当 MNi 数量较少时需要较多的 BN 细胞,这样能在不同处理之间保持统计效力。缺点是当 M N 频率较低时可能需要计数超过 2000 个细胞。我们的实验显示每个重复统计 1000 个 BN 细胞可以得到可信结果。
(7)在观察玻片时首先计数单核细胞、双核、多核、凋亡和坏死细胞频率来确定 ND1 和NDCI。 然后计算双核细胞中的微核、核质桥和核芽数量以确定遗传损伤的比例。
来源:丁香实验