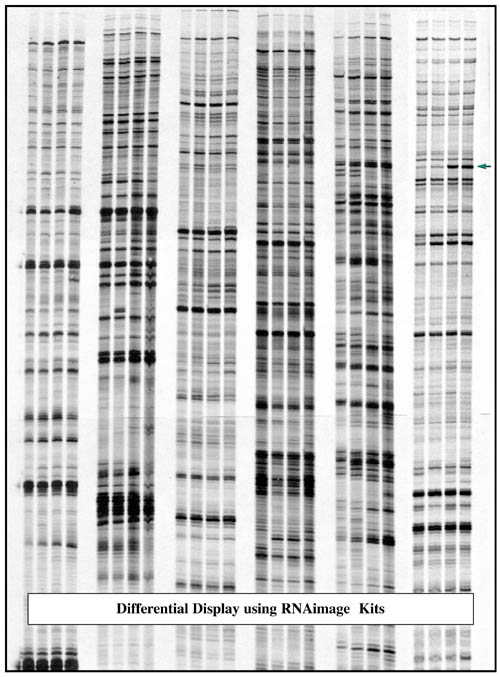
Differential Display of RNAs
互联网
667
The differential display protocol described here is based on the principle described by:
Liang, P. and A.B. Pardee : Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction, Science 257 , 967-971 (1992)
The oligonucleotides we use are kits, contaning 20 decamers, from Operon Technologies Inc. .
![]() I. DNase I treatment of RNA
I. DNase I treatment of RNA
- mix and incubate for 30 min. at 37 °C : 50 µgRNA, 10 µl RNase inhibitor (1 U/µl), 1 µl RNase-free DNaseI (10 U/µl), 5 µl 0.1 M Tris-Cl pH 8.3, 5 µl 0.5 M KCl, 5 µl 15 mM MgCl 2
- phenol extract once
- precipitate with 5 µl NaAcetate and 200 µl 100 % Ethanol
- incubate at least for 30 min at -80 °C
- spin, wash and redissolve in 20 µl H 2ODEPC
- measure RNA concentration and check integrity on a denaturing gel
![]() II. cDNA synthesis
II. cDNA synthesis
- for each RNA set up four reactions (one tube for each degenerate anchored oligo(dT) primer set - T 12MA,T12 MC, T12 MG, T12 MT ; where M is A,C or G)
- dilute DNA-free RNA to 0.1 µg/µl with H2 ODEPC , keep on ice
- set up cDNA synthesis reaction for each degenerate anchored oligo(dT) primer set:
- 4 µl 5 x reverse transcriptase buffer, 2 µl DTT (0.1 M), 1.6 µl 4dNTP mix (250 µM), 200 ng RNA, 2 µl T12 MN primer (10 pmol/µl), with H2 O to 19 µl
- incubate for 5 min at 65 °C and for 10 min at 37 °C
- add 1 µl MMLV reverse transcriptase (200 U/µl)
- incubate for 50 min at 37 °C
- inactivate MMLV RT by incubation for 5 min at 95 °C
- use immediately for PCR amplification or store at -20 °C
![]() III. PCR reaction
III. PCR reaction
- all PCR s should be done in doublets and always run in the same machine (to minimize variations)
- prepare mastermix for all PCR reactions which contain the same T12 MN primer, aliquot 18 µl into each tube and then add the arbitrary primer (= decamer)
- mix for each 20 µl PCR reaction: 9.2 µl H2 O, 2 µl 10 x PCR reaction buffer, 1.6 µl 4dNTP mix (25 µM), 2 µl T12 MN primer, 2 µl cDNA, 0.2 µl Taq DNA polymerase (5 U/µl), 1 µl [a- 33P]dCTP
- aliquot and add 2 µl arbitrary primer (2 pmol/µl)
- run PCR: 40 cycles (94 °C, 30 sec.) (40 °C, 2 min.) (72 °C, 30 sec.), 1 cycle (72 °C, 5 min.)
- store PCR reactions at -20 °C until the gelrun
![]() IV. denaturing gel
IV. denaturing gel
- prepare 6 % denaturing polyacrylamide gel (7 M urea, 1 x TBE), prerun for 30 min at 60 W
- mix 3.5 µl of the PCR reaction with 2 µl formamide loading buffer , incubate 3 min at 95 °C , chill on ice
- load samples (together with a labelled size marker) and run gel until xylene cyanol dye nearly reaches the bottom (3 hours)
- place gel without fixation on Whatman 3MM filter paper and dry on a gel dryer
- autoradiograph for 24 to 48 hours
![]() V. isolation of PCR fragment
V. isolation of PCR fragment
- cut out band of interest with a clean razor blade
- soak in 100 µl H 2O for 10 min at room temperature in a microcentrifuge tube
- boil for 15 min
- spin and take supernatant (containing the DNA) into fresh tube
- precipitate with 10 µl 3 M NaAcetate, 5 µl glycogen (10 mg/ml) , 400 µl 100 % Ethanol
- incubate at -70 °C for at least 30 min
-
spin, wash with 85 % Ethanol and redissolve in 10 µl H 2 O
![]() VI. reamplification
VI. reamplification
- reamplify in a 20 µl PCR reaction (using for each fragment the appropriate T12 MN - arbitrary primer combination)
- mix 4 µl of the isolated DNA , 2 µl 10 x PCR reaction buffer, 1.6 µl 4dNTP mix (250 pmol/µl), 2 µl T12 MN primer, 2 µl arbitrary primer (decamer), 0.2 µl Taq DNA polymerase (5 U/µl)
- use the PCR conditions as under section III.
- run PCR product on a 1.5 % agarose gel and isolate fragment of expected size
- if there is no visible PCR product, take 1 µl of the first reamplification and reamplify again
-
the isolated PCR fragment can be used directly for cycle sequencing , subcloning or
as probe for northern blots