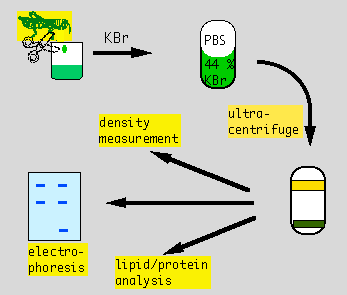
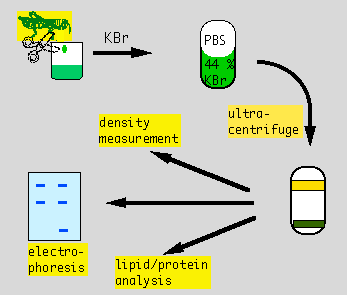
Lipoprotein Analysis Week 2: Electrophoresis
互联网
Lipoprotein Analysis Week 2: Electrophoresis
-
Introduction
SDS polyacrylamide gel electrophoresis (SDS PAGE) will be used to assess the purification process and to determine the apparent molecular weights of the three apoproteins.
SDS-Polyacrylamide gel electrophoresis
Electrophoresis is the process in which charged particles migrate through a solid or liquid matrix in response to application of an electric field. Rate of particle movement is proportional to the charge:mass ratio of the particle and to its frictional resistance. Larger particles move more slowly, and highly charged particles move more quickly. In protein electrophoresis, these factors tend to balance out. Size and charge of a protein determine its electrophoretic mobility. If proteins are separated through a gel matrix with varying pore size, migration depends on the size and shape of the protein. Smaller proteins are retained less, and thus move faster. On the other hand, the net charge of a protein depends on the pH. In native gel electrophoresis, both charge and size determine the migration pattern; in this technique excellent separation can be achieved, but unambiguous information about the protein size cannot be obtained.
However, proteins which have a similar net charge separate nicely according to their size, provided they are of similar shape. For example, globular proteins are retained less than rod-like proteins of the same molecular weight. Experimentally, we can eliminate the influence of charge on protein migration by providing all proteins with extreme negative charges. Sodium dodecyl sulphate (SDS), is an anionic detergent that binds to proteins. Its action is to denature the protein by solubilizing it and effectively "coating" it with a negative charge. The effect before electrophoresis is to block protein interactions with other proteins, polymers such as nucleic acids and lipid, to dissociate multimeric proteins; and to alter folding in protein monomers. SDS not only provides proteins with a strong negative charge, but it also denatures the protein, thus giving each protein a roughly globular shape; differences in protein shape that would affect electrophoretic mobility are eliminated.
The gel matrix most commonly used for proteins separation is polyacrylamide. Polyacrylamide gels are formed when monomeric acrylamide is polymerized by the action of a radical forming agent, ammonium peroxidisulfate (ammonium persulfate) and N,N,N',N'-Tetramethylenediamine (TEMED). The gel forms optimally in the absence of free oxygen, since oxygen is a stable di-radical which can terminate the radical induced polymerization reaction. Since acrylamide polymerizes to long linear products, a cross-linker is required to form a three-dimensional gel. Bisacrylamide serves this function. Pore size within the gel is determined by both, the total acrylamide concentration (% T; = g acrylamide + g bisacrylamide per 100 ml) and the relative concentration of the cross-linker bisacrylamide (% C; = g bisacrylamide per 100 g (acrylamide + bisacrylamide). While pore size decreases with increasing T, small and large C-values yield large pores; the smallest pores are formed in the presence of approx. 5 % C, as seen in this electron micrograph .
Gels can be cast as columns or slabs. For analytical purposes, slabs are much more widely used, since they allow the separation and comparison of multiple samples. In either case, a "stacking" gel is placed on the top of the separating ("running") gel to sharpen the bands before they enter the gel. The electrophoresis buffer and the buffer in the separating gel have a high pH (8.9) and contain glycine. In contrast, the stacking gel buffer has a low pH (6.8) and contains Cl-. At the low pH of the stacking gel, the Cl- in the stacking gel are negatively charged and hence move towards the anode (+), but the glycine entering from the gel buffer has only a very small negative charge (pI of glycine ~ 6). Thus, Cl- moves faster than glycinate, and within the stacking gel a zone of low anion concentration (= low conductivity) forms. This leads to a higher electric field, which accelerates the proteins so that they enter the separating gel as a narrow band at the boundary between the leading Cl- and the trailing glycinate ions. When the protein complexes reach the running gel (pH 9.8), the glycine becomes completely dissociated, and migrates at the same speed as Cl-. You can look at a schematic illustration of this mechanism.
Day 1
Set up SDS-minigel according to the BioRad instructions , as outlined below.
Assembling the Glass Plate Sandwiches
1. Assemble the gel sandwich on a clean surface. Lay the longer rectangular glass plate down first, then place two spacers of equal thickness along the short edges of the rect-angular plate. Next, place the shorter glass plate on top of the spacers so that the bottom ends of the spacers and glass plates are aligned ( Figure ). At this point, the spacers should be sticking up about 5 mm above the long glass plate.2. Loosen the four screws on the clamp assembly and stand it up so that the screws are facing away from you. Firmly grasp the glass plate sandwich with the longer plate facing away from you, and gently slide it into the clamp assembly along the front face of the acrylic pressure plate. The longer glass plate should be against the acrylic pressure plate of the clamp assembly. Tighten the top two screws of the clamp assembly.
3. Place the clamp assembly into the alignment slot so that the clamp screws face away from you. Loosen the top two screws to allow the plates and spacers to settle against the casting stand base. Insert the Mini-PROTEAN II alignment card between the glass plates, in order to position the spacers properly. Gently tighten both pairs of screws.
4. Remove the alignment card. Pull the completed sandwich from the alignment slot. Check that the plates and spacers are flush at the bottom. If not, realign the sandwich as in steps 1-3.
5. Using the leveling bubble, level the casting stand with the alignment slot facing you. Check to see that the removable gray silicone gaskets are clean and free of residual acry-lamide to insure a good seal. Place the silicone rubber gaskets on top of the red foam pads of the casting stand slots.
6. Transfer the clamp assembly to one of the casting slots in the casting stand. If two gels are to be cast, place the clamp assembly on the side opposite the alignment slot to make aligning the next sandwich easier.
7. Attach the sandwich in the following way: Butt the acrylic pressure plate against the wall of the casting slot at the bottom, so the glass plates rest on the rubber gasket. Snap the acrylic plate underneath the overhang of the casting slot by pushing with the white portions of the clamps (see Figure ). Do not push against the glass plates or spacers. This could break the plate.
Note: It is especially important to assure that the rubber gasket is placed correctly (with notch facing glass plate), and that the bottom is aligned exactly to give a smooth seal. It is recommended to fill the assembled cassette first partially with water, marking the meniscus with a felt tip pen. If no leakage is detected in 5 minutes, pour out the water and remove the residual water by inserting a filter paper. You can then begin to pour the gel.
Preparation of separating gel
To prepare 20 ml of homogeneous gel (this amount is for two mini gels) with the concentration given below, pipette out the amounts shown (with the exception of TEMED and the SDS solution) in the following table into a 250 ml side armed Erlenmeyer flask.
Note: Degas the mixture before adding TEMED and SDS.
<center> <table> <tbody> <tr> <td> <font><font><font><font>Stock</font> </font></font></font></td> <td> <font><font><font><font>final conc.</font> </font></font></font></td> <td> <font><font><font><font>Amount to use</font> </font></font></font></td> </tr> <tr> <td> <font><font><font><font>1.5 M Tris-HCl</font> </font></font></font></td> <td> <font><font><font><font>0.375M </font> </font></font></font></td> <td> <font><font><font><font>5 ml</font> </font></font></font></td> </tr> <tr> <td> <font><font><font><font><font><b>30</b> % Acryl:Biundefined </font> </font> </font></font></font></td> <td> <font><font><font><font>10 % </font> </font></font></font></td> <td> <font><font><font><font>7.7 ml</font> </font></font></font></td> </tr> <tr> <td> <font><font><font><font>10% SDS</font> </font></font></font></td> <td> <font><font><font><font>0.1% </font> </font></font></font></td> <td> <font><font><font><font>0.2 ml</font> </font></font></font></td> </tr> <tr> <td> <font><font><font><font>10% APS </font> </font></font></font></td> <td> <font><font><font><font>0.05% </font> </font></font></font></td> <td> <font><font><font><font>100 µl</font> </font></font></font></td> </tr> <tr> <td> <font><font><font><font>H2O </font> </font></font></font></td> <td> </td> <td> <font><font><font><font>8.0 ml</font> </font></font></font></td> </tr> <tr> <td> <font><font><font><font>TEMED </font> </font></font></font></td> <td> <font><font><font><font>0.0005% </font> </font></font></font></td> <td> <font><font><font><font>10 µl</font> </font></font></font></td> </tr> </tbody> </table> <font><font><font><font>~undefineduse the 30 % acrylamide stock solution!</font> </font></font></font></center>1. Use a plastic 10 ml pipette to pour the gel in to plates. Pour resolving gel up to ~ 2 cm from top.
2. To avoid exposure to air, carefully layer water on top of the resolving gel. Leave the gel to polymerize. A sharp line between water layer and gel indicates completion of polymerization. While waiting for the gel to polymerize you can start preparing the stacking gel.
Preparation of stacking gel
Prepare a 7.5 ml of 3% stacking gel in a small beaker using the following amounts of appropriate reagents.
<center> <table> <tbody> <tr> <td> <font><font><font><font><font>Stock</font> </font></font></font></font></td> <td> <font><font><font><font><font>final conc.</font> </font></font></font></font></td> <td> <font><font><font><font><font>Amount to use</font> </font></font></font></font></td> </tr> <tr> <td> <font><font><font><font><font>0.5 M Tris-HCl</font> </font></font></font></font></td> <td> <font><font><font><font><font>0.125M </font> </font></font></font></font></td> <td> <font><font><font><font><font>1.88 ml</font> </font></font></font></font></td> </tr> <tr> <td> <font><font><font><font><font><font><b>10</b> % Acryl:Biundefined </font> </font> </font></font></font></font></td> <td> <font><font><font><font><font>3% </font> </font></font></font></font></td> <td> <font><font><font><font><font>2.25 ml</font> </font></font></font></font></td> </tr> <tr> <td> <font><font><font><font><font>10% SDS</font> </font></font></font></font></td> <td> <font><font><font><font><font>0.1% </font> </font></font></font></font></td> <td> <font><font><font><font><font>0.075 ml</font> </font></font></font></font></td> </tr> <tr> <td> <font><font><font><font><font>10% APS </font> </font></font></font></font></td> <td> <font><font><font><font><font>0.1% </font> </font></font></font></font></td> <td> <font><font><font><font><font>0.10 ml</font> </font></font></font></font></td> </tr> <tr> <td> <font><font><font><font><font>H2O </font> </font></font></font></font></td> <td> </td> <td> <font><font><font><font><font>3.19 ml</font> </font></font></font></font></td> </tr> <tr> <td> <font><font><font><font><font>TEMED </font> </font></font></font></font></td> <td> <font><font><font><font><font>0.00067% </font> </font></font></font></font></td> <td> <font><font><font><font><font>10 µl</font> </font></font></font></font></td> </tr> </tbody> </table> </center> <center> <font><font><font><font><font>~undefinedUse the 10 % stock solution!</font> </font></font></font></font></center>3. When the polymerization of resolving gel is complete, decant the layer of water. Dry excess water using Kim-wipes.
4. Pour the stacking gel using a pasture pipette. Insert the comb gently. Leave to polymerize until gel turns milky (at least 30 min.).
Assembling the Upper Buffer Chamber
Note: To insure a leakproof seal, make sure the gray U-shaped inner cooling core gaskets are clean. Inspect the gasket for small cuts that could result in an upper buffer leak. There are two sides to this gasket. Make sure that the side with the notch is exposed for contact with the gel sandwich.
1. Release the clamp assemblies/gel sandwiches from the casting stand.
2. Lay the inner cooling core down flat on a lab bench. With the glass plates of the gel sandwich facing the cooling core (and the clamp screws facing out), carefully slide the clamp assembly wedges underneath the locator slots on the inner cooling core until the inner glass plate of the gel sandwich butts up against the notch in the U-shaped gasket (Figure 5.1).
Note: Lubricating the raised portions of the U-shaped gasket with a drop of running buffer or water helps the glass plate sandwich slide in properly.
While pushing the clamp assembly slightly up toward the top of the locator slots, snap the clamp assembly fully onto the cooling core by pressing at the bottom of the clamp assembly until the cooling core latch engages each side of the clamp assembly. (Do not pull out on cooling core latch at the same time.)Electrophoresis
1. Remove the 10 dialyzed fractions collected after the density gradient centrifugation and the pooled lipophorin sample from the cold room. Remove 50 µl each and place into a pre-labeled Eppendorf tubes. Label the tube at the lid with a waterproof pen; otherwise you won't be able to identify your samples!
2. Add 25 µl sample buffer to each fraction and close lids. Together with one tube of molecular weight markers, place in the sample holder and boil for 2 min.
<center> </center>
Use the following molecular weights:3. In the meantime, remove the comb from the gel. Mark the wells with a felt pen. This will enable you to see the wells clearly when the running buffer is poured into the upper chamber. Assemble the upper buffer chamber.
5. Dilute 60 ml of 5x stock of running buffer with 240 ml of dist. water. Pour carefully into the upper buffer chamber until the wells are covered. Pour the rest of the buffer into lower buffer chamber along the walls of the container. Make sure no air bubbles are trapped under the gels. If present, you can remove air bubbles using a wire bent at the tip.
6. Have two standard molecular weight markers (high and low range) ready. It is not necessary to add sample buffer to the molecular weight markers since it has already been added. These should be loaded into the two corner wells.
7. Using a clean Hamilton syringe load the samples in to the wells. Load 10 µl of each sample. For the pooled lipophorin sample, load three different amounts to assure a good banding pattern for densitomentric analysis: e.g., 2 µl, 5 µl, 15 µl. Rinse the syringe well with dist. water after each sample.
8. Place the cover and attach the power supply. Turn on the power and set run voltage to 125 volts. Approximate run time is about 1 h.
9. while the the gel is running, drop the tube with the lipophorin in liquid nitrogen.
10. Place a parafilm on top of the tube and pierce the film with a needle.
11. Leave in a lyophilizer jar and attach to the lyophilizer.
12. Freeze dry (lyophilize) overnight or longer.
13. Once the electrophoresis run is complete (when the marker dye reaches approximately 1 cm from the bottom of the gel), turn the power off. Remove the gels from the upper buffer chamber. Lay the inner cooling core on its side and remove the clamp assembly by pushing down on both sides of the cooling core latch and up on the clamps until the clamp assembly is released. Slide the clamp assembly away from the cooling core. Open the scres and remove the gel sandwich.
14. Prop open the glass plate. Remove the gel carefully and leave in the stain. Stain overnight.
Day 2 (you will need to come 2 or 3 times for 10 minutes)
1. Decant used stain into the bottle assigned. Pour destain and leave on shaker for 2-3 h or until bands are visible in a lighter background.
2. The gels can be stored in dilute destain solution (destain:water 1:1) in a covered petri dish, sealed with parafilm.