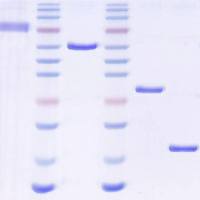
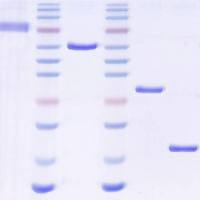
The ribonuclease protection assay (RPA)
互联网
The ribonuclease protection assay (RPA) is a highly sensitive and specific method for the detection of mRNA species. The assay was made possible by the discovery and characterization of DNA-dependant RNA polymerases from the bacteriophages SP6, T7 and T3, and the elucidation of their cognate promoter sequences. These polymerases are ideal for the synthesis of high-specific-activity RNA probes from DNA templates because these polymerases exhibit a high degree of fidelity for their promoters, polymerize RNA at a very high rate, efficiently transcribe long segments, and do not require high concentrations of rNTPs. Thus a cDNA fragment of interest can be subcloned into a plasmid that contains bacteriophage promoters, and the construct can then be used as a template for synthesis or radiolabeled anti-sense RNA probes.
Standard RPA Procedure
In all steps of the protocol, standard precautions should be used to avoid RNase contamination and exposure of personnel to radioactivity. Typically, the probe synthesis is performed during the afternoon Day 1, hybridizations are incubated overnight, and RNase treatments and gel electrophoresis are performed early on Day 2.
Probe Synthesis:
1.
Bring the [a-32P]UTP, GACU nucleotide pool, DTT, 5X transcription buffer, and RPA template set to RT. For each probe synthesis, add the following in order to a 1.5 ml Eppendorf tube:
1 µl RNasin®
1 µl GACU pool
2 µl DTT
4 µl 5X transcription buffer
1 µl RPA Template Set
10 µl [a-32P]UTP
1 µl T7 RNA polymerase (Keep at -20°C until use, return to -20°C immediately).
Mix by gentle pipetting or flicking and quick spin in a microfuge. Incubate at 37°C for 1 hour.
2.
Terminate the reaction by adding 2 µl of DNase. Mix by gentle flicking and quick spin in a microfuge. Incubate at 37°C for 30 minutes.
3.
Add the following reagents (in order) to each 1.5 ml Eppendorf tube:
26 µl 20 mM EDTA
25 µl Tris-saturated phenol
25 µl chloroform:isoamyl alcohol (50:1)
2 µl yeast tRNA
Mix by vortexing into an emulsion and spin in a microfuge for 5 minutes at RT.
4.
Transfer the upper aqueous phase to a new 1.5 ml Eppendorf tube and add 50 µl chloroform:isoamyl alcohol (50:1). Mix by vortexing, then spin in a microfuge for 2 minutes at RT.
5.
Transfer the upper aqueous phase to a new 1.5 ml Eppendorf tube and add 50 µl 4 M ammonium acetate and 250 µl ice cold 100% ethanol. Invert the tube![]() to mix and incubate for 30 minutes at -70°C. Spin in a microfuge for 15 minutes at 4°C.
to mix and incubate for 30 minutes at -70°C. Spin in a microfuge for 15 minutes at 4°C.
6.
Carefully remove the supernatant and add 100 µl of ice cold 90% ethanol to the pellet. Spin in a microfuge for 5 minutes at 4°C.
7.
Carefully remove the supernatant and air dry the pellets for 5 to 10 minutes (do not dry in a vacuum evaporator centrifuge). Add 50 µl of hybridization buffer and solubilize the pellet by gently vortexing for 20 seconds and quick spin on microfuge.
8.
Quantitate duplicate 1 µl samples in the scintillation counter. Expect a maximum yield of 1-3 x 106 Cherenkov counts / µl (measurement of cpm / µl without the presence of scintillation fluid) with an acceptable lower limit of 3 x 105 Cherenkov counts / µl. Store the probe at -20°C until needed. Generally, the probe can be used for two successive overnight hybridizations at most.
RNA Preparation & Hybridization:
9.
For the best results, use procedures that generate total RNA of high quality and purity. RNA should be stored in RNase-free water at -70°C. Add the desired amount of target RNA (generally 1-20 µg) to 1.5 ml Eppendorf tubes and include a tube that contains yeast tRNA as a background control. In general, 20-24 total sample tubes are an easily manageable number for each RPA setup. With the Pharmingen control RNA, 2 µl volume (i.e., 2 µg) is recommended.
10.
If RNA has been stored in water, freeze the samples for 15 minutes at -70°C. Dry completely (~1 hour) in a vacuum evaporator centrifuge (no heat). Likewise, RNA can be precipitated prior to the addition of hybridization buffer as in Step 5.
11.
Add 8 µl of hybridization buffer to each sample. Solubilize the RNA by gently vortexing for 3-4 minutes and quick spin in the microfuge.
12.
Dilute the probe from Step 8 with hybridization buffer to the appropriate concentration: Add 2 µl of diluted probe to each RNA sample and mix by pipetting. Add a drop of mineral oil to each tube and quick spin in the microfuge.
13.
Place the samples in a heat block pre-warmed to 90°C. Immediately turn the temperature to 56°C (allowing the temperature to ramp down slowly) and incubate for 12-16 hours. Turn the heat block to 37°C for 15 minutes prior to the RNase treatments, again allowing the temperature to ramp down slowly. All incubations may also be carried out in a water bath.
RNase Treatments:
14.
Prepare the RNase cocktail (per 20 samples)
2.5 ml RNase buffer
6 µl RNase A + T1 mix
Remove the RNA samples from the heat block and pipet 100 µl of the RNase cocktail underneath the oil into the aqueous layer (bubble). Spin in microfuge for 10 seconds and incubate for 45 minutes at 30°C.
15.
Before the RNase digestion is completed, prepare the Proteinase K cocktail (per 20 samples):
390 µl Proteinase K buffer
30 µl Proteinase K
30 µl yeast tRNA
Mix and add 18 µl aliquots of the cocktail to new Eppendorf tubes.
16.
Using a pipettor, extract the RNase digests from underneath the oil (try to avoid the oil) and transfer to the tubes containing the Proteinase K solution. Quick vortex, quick spin in the microfuge, and incubate for 15 minutes at 37°C.
17.
Add 65 µl Tris-saturated phenol and 65 µl chloroform:isoamyl alcohol (50:1). Vortex into an emulsion and spin in the microfuge for 5 minutes at RT.
18.
Carefully extract the upper aqueous phase (set the pipettor at 120 µl and totally avoid the organic interface) and transfer to a new tube. Add 120 µl 4 M ammonium acetate and 650 µl ice cold 100% ethanol. Mix by inverting the tubes and incubate for 30 minutes at -70°C. Spin in the microfuge for 5 minutes at 4°C.
19.
Carefully remove the supernatant and add 100 µl ice cold 90% ethanol. Spin in the microfuge for 5 minutes at 4°C.
20.
Carefully remove the supernatant and air dry the pellet (do not dry in a vacuum evaporator centrifuge). Add 5 µl of 1X loading buffer, vortex for 2-3 minutes, and quick spin in the microfuge. Prior to loading the samples on the gel, heat the samples for 3 minutes at 90°C and then place them immediately in an ice bath.
Gel Resolution of Protected Probes:
21.
Clean a set of gel plates (> 40 cm in length) thoroughly with water followed by ethanol. Siliconize the short plate and clean again. Assemble the gel mold (0.4 mm spacers).
22.
Combine the following to give a final concentration of 5% acrylamide:
74.5 ml acrylamide solution (final 19:1 acrylamide/bis):
8.85 mls of 40% acrylamide
9.31 mls of 2% bis acrylamide
7.45 mls of 10x TBE
35.82 g of Urea
QS to 74.5 ml with dH2O
450 µl ammonium persulfate (10%)
60 µl TEMED
Pour immediately into the gel mold, remove any air bubbles, and add an appropriate comb (e.g., 5 mm well width). Use of a sharks tooth comb is not recommended.
23.
After polymerization (~1 hour), remove the comb and flush the wells thoroughly with 0.5X TBE. Place each gel in a vertical rig (use a gel set up that has a heat dispenser) and prerun at 40 watts constant power for ~ 45 minutes, with 0.5X TBE as the running buffer. Gel temperature should be 50°C.
24.
Flush the wells again with 0.5X TBE and load the samples (from Step 20). Also load a dilution of the probe set in loading buffer (typically 1000-2000 cpm/lane) to server as size markers. Run the gel at 50 watts constant power until the leading edge of the Bromophenol Blue (BP![]() (front dye) reaches 30 cm.
(front dye) reaches 30 cm.
25.
Disassemble the gel mold, remove the short plate, and absorb the gel to filter paper. Cover the gel with Saran wrap and layer between two additional pieces of filter paper. Place in the gel dryer vacuum for ~ 1 hour at 80°C. Place the dried gel on film (Kodak X-AR) in a cassette with an intensifying screen and develop at -70°C (Exposure times will vary depending on application). Alternatively, radioactivity can be quantified by phosphorimaging or other equivalent instruments.
26.
With the undigested probes as markers, plot a standard curve on a semi-log graph paper, of migration distance versus log nucleotide length. Use this curve to establish the identity of "RNase-protected" bands in the experimental samples. Note that the probe lengths are greater than the "protected" fragment lengths, this is due to the presence of flanking sequences in the probes that are derived from the plasmid and do not hybridize with target mRNA.