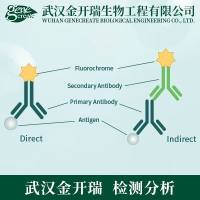
RNA实验方法
互联网
2681
1. A Hoeffer/LKB Transphor unit is recommended for Northern blots. The Bio-Rad unit has been known to melt when incorrect voltage was applied.
The recommended transfer membrane for very small RNA was 0.1 � nylon (Schleicher & Schuell Nytran or ICN Biotrans) but this material is no longer available. RNA that is < 110 nucleotides will tend to go through a 0.2 � membrane. Try Schleicher & Schuell nitrocellulose membranes (BA79/0.1 � and BA75/0.05 �)? Nylon (neutral or charged) membranes are rated for denatured > 50 bp DNA fragments, require alkaline transfer conditions and may have higher backgrounds.
Hoeffer sponges are 6?x 9?and the gel must be cut to fit these dimensions.
The Hoeffer gel box can blot 4 gels simultaneously if additional cassettes are purchased.
The Hoeffer unit generates more heat than the BioRad unit.
BioRad sponges are 6 1/8?x 8 3/8?and the gel must be cut to fit these dimensions. The BioRad box is limited to a single gel.
Polyacrylamide/TBE gels ranging from 4 to 8% have been successfully used, with or without urea. It is not necessary to presoak the gel to remove urea prior to blotting. If the gel is severely overloaded, some RNA will transfer to a second layer of nylon.
2. Water and buffers do not need to be DEPC-treated if they do not come from carboys. Carboy spouts tend to be a source of mildew RNases that are resistant to DEPC.
3. Prep a clean tray large enough to hold the blot cassette and soak in 0.1% DEPC or 3% hydrogen peroxide for 10 min at room temperature. Cover the pan during the soak.
4. Prep 5000 mL 1x TAE5 for the Hoeffer unit or 3300 mL 1x TAE5 for the BioRad unit. Pre-chill the buffer on ice in the coldroom.
5. Cut nylon or nitrocellulose and 2 pieces of Whatman 3MM to required dimensions while wearing gloves to prevent finger oils from coming into contact with membrane. Clip one corner to indicate orientation.
6. Drain 0.1% DEPC or 3% hydrogen peroxide from the tray. Rinse with a trace of 1x TAE5 . Add remaining portion of 1 liter 1x TAE5 to tray. Add plastic gel cassette and sponges. Squeeze the air out of the sponges. Soak the membrane briefly by laying it on the sponges.
7. Open the gel plates. Apply membrane to gel and gently brush out air bubbles. Dip one piece of 3MM very briefly in 1x TAE5 and lay on top of membrane. Remove air bubbles. Lay glass plate on 3MM and flip gel over. Remove the other plate (this seems to be difficult) and prewet the remaining piece of 3MM. Lay this on top of the gel. Remove air bubbles. Move to plastic cassette and assemble holder.
8. Place the membrane side of the gel cassette towards the red electrode. Add prechilled 1x TAE5 buffer to cover. Use the 1x TAE5 buffer in the presoak tray as part of the required buffer.
9. Careful! Electrode plugs on the BioRad Transblot will corrode when wet. Keep the buffer below the electrode plugs.
10. Place a magnetic stir bar in the bottom of the box. Move box to 4� coldroom. Buffer must be stirred during blotting run. Set magnetic stirrer at speed 6.
11. Hoeffer/LKB Transphor 10 v/30 min then 40 v/2 hours or 20 v/overnight
BioRad Transblot 20 v overnight. Do not use the 40 volt protocol with the BioRad unit.
12. UV fix the wet membrane (leave it on the wet 3MM support) with 120,000 �oules UV via the UV crosslinker. Alternatively, use a Sylvania germicidal UV lamp exactly 12 cm from the bulb for exactly 2 min. Caution: nitrocellulose is highly flammable. Air dry the membrane. Place a single membrane in a hybridization bag. Seal the bag. Cut one corner and add prehyb solution at 5 mL/100 cm2 membrane. Incubate in shaking waterbath for 3 hours at 42�. Cut a corner of the bag and remove all prehyb. Add hybridization solution at 5 mL/100 cm2 membrane.
13. Add 1 x 107 cpm 32 P-labeled probe to 300 � reserved hyb mix and boil for 3-5 min. Transfer the probe to ice and then add the probe to the hybridization bag within 20 min. Seal the bag. Rinse the bag in the sink to remove prehyb or hyb mix from the outside of the bag, as these will support the growth of bacteria in the waterbath. Incubate the hyb solution in a shaking waterbath overnight at 55�. Alternatively, a rotisserie apparatus can be used instead of hybridization bags and shaking water baths.
13. Prehybridization mix (add in order listed at 5 mL/100 cm2 membrane)
5x Denhardt�, 5x SSC, 50 mM NaPO4 , pH 6.7, 50% deionized formamide250 �/mL sheared salmon sperm DNA, 1% dextran sulfate MW 5000 and 0.1% SDS
Prep the hyb mix and prehyb mix fresh each time. The hyb mix can sit at room temperature during the 3 hour prehyb step.
14. Hybridization mix (add in order listed at 5 mL/100 cm2 membrane)
14. Hybridization mix (add in order listed at 5 mL/100 cm2 membrane):
1x Denhardt�, 5x SSC, 20 mM NaPO4 , pH 6.7, 50% deionized formamide, 100 �/mL sheared salmon sperm DNA, 5% dextran sulfate MW 5000, and 0.1% SDS
15. Wash square Pyrex dish that will fit in shaking waterbath. Soak in 3% hydrogen peroxide or 0.1% DEPC for 10 min.
16. High Stringency Washes 1, 2 and 3
Wash 1 2x SSC: 80 mL 25x SSC, pH 7.0, 910 mL Type I water, 10 mL 10% SDS
Wash 2 1x SSC: 40 mL 25x SSC, pH 7.0, 940 mL Type I water, 10 mL 10% SDS
Wash 3 0.5x SSC: 20 mL 25x SSC, pH 7.0, 970 mL Type I water, 10 mL 10% SDS
17. Discard the probe and hybridization solution in 32 P liquid waste. Rinse the bag with approximately 30 mL wash 1 and discard the wash in 32 P liquid waste. Discard the 0.1% DEPC in the tray and add approximately 100 mL wash 1 to the dish. Transfer the membrane to the dish and cover with Saran. Shake gently at room temperature for 10 min. Discard wash in 32 P waste. Microwave the remaining wash for 1 min to prewarm slightly and add to the Pyrex dish containing the probed membrane. Incubate at 65� for 30 min. Washes may be performed in the hybridization bag, but may result in higher background. Discard wash 1 and add 1000 mL prewarmed wash 2 and incubate at 65� for 30 min. Discard wash 2 and add 1000 mL prewarmed wash 3 and incubate at 65� for 30 min.
18. Air dry the filter on 3MM paper and wrap with Saran. Expose the membrane as desired.
Do not let the membrane dry out if reprobing is required. Place the blot in a clear plastic sheet protector and seal it closed with tape for chemiluminescent exposure. For storage, seal the blot in a water-tight seal-a-meal pouch and store at ?0�. Note that only SaranWrap is water-tight. Other plastic wraps are water-permeable and allow blots to dry out (and allow water from wet gels to contact autoradiography film, causing black spots).
Neutral membranes have an equal and uniform distribution of charges. Positively charged membranes have a preponderance of positive charges across the matrix. They will hold the same amount of nucleic acid. Positively charged membranes have stronger retention: the target remains bound longer. UV crosslinking forms a covalent bond between nucleic acids and the positive charges of the membrane. Fixation by baking creates a hydrophobic interaction between the nucleic acids and the membrane. If stripping and reprobing is required, it is better to UV crosslink to a positively charged membrane in order to preserve the integrity of the target during the stripping step.
Quality membranes do not have a correct side. The charges are equally distributed on both sides.
Nominally, 0.22 � pore membranes have greater surface area than 0.45 � membranes. They also require better blocking. Consistent pore size is critical. For Northern blots of small RNAs (~ 100 nucleotides), 0.1 � membranes are recommended.
Particulates in unfiltered buffer, skin oils, nonspecific antibody binding, addition of too much probe, inadequate post-hybridization washing, improper wash temperature, and other factors can create background fog or spots on membranes.
Blocking can reduce background. Non-specific proteins block certain electrostatic interactions which may attract proteins randomly. Milk proteins, BSA, or commercial blocking agents are typically used.
1. A Hoeffer/LKB Transphor unit is recommended for Northern blots. The Bio-Rad unit has been known to melt when incorrect voltage was applied.
The recommended transfer membrane for very small RNA was 0.1 � nylon (Schleicher & Schuell Nytran or ICN Biotrans) but this material is no longer available. RNA that is < 110 nucleotides will tend to go through a 0.2 � membrane. Try Schleicher & Schuell nitrocellulose membranes (BA79/0.1 � and BA75/0.05 �)? Nylon (neutral or charged) membranes are rated for denatured > 50 bp DNA fragments, require alkaline transfer conditions and may have higher backgrounds.
Hoeffer sponges are 6?x 9?and the gel must be cut to fit these dimensions.
The Hoeffer gel box can blot 4 gels simultaneously if additional cassettes are purchased.
The Hoeffer unit generates more heat than the BioRad unit.
BioRad sponges are 6 1/8?x 8 3/8?and the gel must be cut to fit these dimensions. The BioRad box is limited to a single gel.
Polyacrylamide/TBE gels ranging from 4 to 8% have been successfully used, with or without urea. It is not necessary to presoak the gel to remove urea prior to blotting. If the gel is severely overloaded, some RNA will transfer to a second layer of nylon.
2. Water and buffers do not need to be DEPC-treated if they do not come from carboys. Carboy spouts tend to be a source of mildew RNases that are resistant to DEPC.
3. Prep a clean tray large enough to hold the blot cassette and soak in 0.1% DEPC or 3% hydrogen peroxide for 10 min at room temperature. Cover the pan during the soak.
4. Prep 5000 mL 1x TAE5 for the Hoeffer unit or 3300 mL 1x TAE5 for the BioRad unit. Pre-chill the buffer on ice in the coldroom.
5. Cut nylon or nitrocellulose and 2 pieces of Whatman 3MM to required dimensions while wearing gloves to prevent finger oils from coming into contact with membrane. Clip one corner to indicate orientation.
6. Drain 0.1% DEPC or 3% hydrogen peroxide from the tray. Rinse with a trace of 1x TAE5 . Add remaining portion of 1 liter 1x TAE5 to tray. Add plastic gel cassette and sponges. Squeeze the air out of the sponges. Soak the membrane briefly by laying it on the sponges.
7. Open the gel plates. Apply membrane to gel and gently brush out air bubbles. Dip one piece of 3MM very briefly in 1x TAE5 and lay on top of membrane. Remove air bubbles. Lay glass plate on 3MM and flip gel over. Remove the other plate (this seems to be difficult) and prewet the remaining piece of 3MM. Lay this on top of the gel. Remove air bubbles. Move to plastic cassette and assemble holder.
8. Place the membrane side of the gel cassette towards the red electrode. Add prechilled 1x TAE5 buffer to cover. Use the 1x TAE5 buffer in the presoak tray as part of the required buffer.
9. Careful! Electrode plugs on the BioRad Transblot will corrode when wet. Keep the buffer below the electrode plugs.
10. Place a magnetic stir bar in the bottom of the box. Move box to 4� coldroom. Buffer must be stirred during blotting run. Set magnetic stirrer at speed 6.
11. Hoeffer/LKB Transphor 10 v/30 min then 40 v/2 hours or 20 v/overnight
BioRad Transblot 20 v overnight. Do not use the 40 volt protocol with the BioRad unit.
12. UV fix the wet membrane (leave it on the wet 3MM support) with 120,000 �oules UV via the UV crosslinker. Alternatively, use a Sylvania germicidal UV lamp exactly 12 cm from the bulb for exactly 2 min. Caution: nitrocellulose is highly flammable. Air dry the membrane. Place a single membrane in a hybridization bag. Seal the bag. Cut one corner and add prehyb solution at 5 mL/100 cm2 membrane. Incubate in shaking waterbath for 3 hours at 42�. Cut a corner of the bag and remove all prehyb. Add hybridization solution at 5 mL/100 cm2 membrane.
13. Add 1 x 107 cpm 32 P-labeled probe to 300 � reserved hyb mix and boil for 3-5 min. Transfer the probe to ice and then add the probe to the hybridization bag within 20 min. Seal the bag. Rinse the bag in the sink to remove prehyb or hyb mix from the outside of the bag, as these will support the growth of bacteria in the waterbath. Incubate the hyb solution in a shaking waterbath overnight at 55�. Alternatively, a rotisserie apparatus can be used instead of hybridization bags and shaking water baths.
13. Prehybridization mix (add in order listed at 5 mL/100 cm2 membrane)
5x Denhardt�, 5x SSC, 50 mM NaPO4 , pH 6.7, 50% deionized formamide250 �/mL sheared salmon sperm DNA, 1% dextran sulfate MW 5000 and 0.1% SDS
Prep the hyb mix and prehyb mix fresh each time. The hyb mix can sit at room temperature during the 3 hour prehyb step.
14. Hybridization mix (add in order listed at 5 mL/100 cm2 membrane)
14. Hybridization mix (add in order listed at 5 mL/100 cm2 membrane):
1x Denhardt�, 5x SSC, 20 mM NaPO4 , pH 6.7, 50% deionized formamide, 100 �/mL sheared salmon sperm DNA, 5% dextran sulfate MW 5000, and 0.1% SDS
15. Wash square Pyrex dish that will fit in shaking waterbath. Soak in 3% hydrogen peroxide or 0.1% DEPC for 10 min.
16. High Stringency Washes 1, 2 and 3
Wash 1 2x SSC: 80 mL 25x SSC, pH 7.0, 910 mL Type I water, 10 mL 10% SDS
Wash 2 1x SSC: 40 mL 25x SSC, pH 7.0, 940 mL Type I water, 10 mL 10% SDS
Wash 3 0.5x SSC: 20 mL 25x SSC, pH 7.0, 970 mL Type I water, 10 mL 10% SDS
17. Discard the probe and hybridization solution in 32 P liquid waste. Rinse the bag with approximately 30 mL wash 1 and discard the wash in 32 P liquid waste. Discard the 0.1% DEPC in the tray and add approximately 100 mL wash 1 to the dish. Transfer the membrane to the dish and cover with Saran. Shake gently at room temperature for 10 min. Discard wash in 32 P waste. Microwave the remaining wash for 1 min to prewarm slightly and add to the Pyrex dish containing the probed membrane. Incubate at 65� for 30 min. Washes may be performed in the hybridization bag, but may result in higher background. Discard wash 1 and add 1000 mL prewarmed wash 2 and incubate at 65� for 30 min. Discard wash 2 and add 1000 mL prewarmed wash 3 and incubate at 65� for 30 min.
18. Air dry the filter on 3MM paper and wrap with Saran. Expose the membrane as desired.
Do not let the membrane dry out if reprobing is required. Place the blot in a clear plastic sheet protector and seal it closed with tape for chemiluminescent exposure. For storage, seal the blot in a water-tight seal-a-meal pouch and store at ?0�. Note that only SaranWrap is water-tight. Other plastic wraps are water-permeable and allow blots to dry out (and allow water from wet gels to contact autoradiography film, causing black spots).
Neutral membranes have an equal and uniform distribution of charges. Positively charged membranes have a preponderance of positive charges across the matrix. They will hold the same amount of nucleic acid. Positively charged membranes have stronger retention: the target remains bound longer. UV crosslinking forms a covalent bond between nucleic acids and the positive charges of the membrane. Fixation by baking creates a hydrophobic interaction between the nucleic acids and the membrane. If stripping and reprobing is required, it is better to UV crosslink to a positively charged membrane in order to preserve the integrity of the target during the stripping step.
Quality membranes do not have a correct side. The charges are equally distributed on both sides.
Nominally, 0.22 � pore membranes have greater surface area than 0.45 � membranes. They also require better blocking. Consistent pore size is critical. For Northern blots of small RNAs (~ 100 nucleotides), 0.1 � membranes are recommended.
Particulates in unfiltered buffer, skin oils, nonspecific antibody binding, addition of too much probe, inadequate post-hybridization washing, improper wash temperature, and other factors can create background fog or spots on membranes.
Blocking can reduce background. Non-specific proteins block certain electrostatic interactions which may attract proteins randomly. Milk proteins, BSA, or commercial blocking agents are typically used.
The recommended transfer membrane for very small RNA was 0.1 � nylon (Schleicher & Schuell Nytran or ICN Biotrans) but this material is no longer available. RNA that is < 110 nucleotides will tend to go through a 0.2 � membrane. Try Schleicher & Schuell nitrocellulose membranes (BA79/0.1 � and BA75/0.05 �)? Nylon (neutral or charged) membranes are rated for denatured > 50 bp DNA fragments, require alkaline transfer conditions and may have higher backgrounds.
Hoeffer sponges are 6?x 9?and the gel must be cut to fit these dimensions.
The Hoeffer gel box can blot 4 gels simultaneously if additional cassettes are purchased.
The Hoeffer unit generates more heat than the BioRad unit.
BioRad sponges are 6 1/8?x 8 3/8?and the gel must be cut to fit these dimensions. The BioRad box is limited to a single gel.
Polyacrylamide/TBE gels ranging from 4 to 8% have been successfully used, with or without urea. It is not necessary to presoak the gel to remove urea prior to blotting. If the gel is severely overloaded, some RNA will transfer to a second layer of nylon.
2. Water and buffers do not need to be DEPC-treated if they do not come from carboys. Carboy spouts tend to be a source of mildew RNases that are resistant to DEPC.
3. Prep a clean tray large enough to hold the blot cassette and soak in 0.1% DEPC or 3% hydrogen peroxide for 10 min at room temperature. Cover the pan during the soak.
4. Prep 5000 mL 1x TAE5 for the Hoeffer unit or 3300 mL 1x TAE5 for the BioRad unit. Pre-chill the buffer on ice in the coldroom.
5. Cut nylon or nitrocellulose and 2 pieces of Whatman 3MM to required dimensions while wearing gloves to prevent finger oils from coming into contact with membrane. Clip one corner to indicate orientation.
6. Drain 0.1% DEPC or 3% hydrogen peroxide from the tray. Rinse with a trace of 1x TAE5 . Add remaining portion of 1 liter 1x TAE5 to tray. Add plastic gel cassette and sponges. Squeeze the air out of the sponges. Soak the membrane briefly by laying it on the sponges.
7. Open the gel plates. Apply membrane to gel and gently brush out air bubbles. Dip one piece of 3MM very briefly in 1x TAE5 and lay on top of membrane. Remove air bubbles. Lay glass plate on 3MM and flip gel over. Remove the other plate (this seems to be difficult) and prewet the remaining piece of 3MM. Lay this on top of the gel. Remove air bubbles. Move to plastic cassette and assemble holder.
8. Place the membrane side of the gel cassette towards the red electrode. Add prechilled 1x TAE5 buffer to cover. Use the 1x TAE5 buffer in the presoak tray as part of the required buffer.
9. Careful! Electrode plugs on the BioRad Transblot will corrode when wet. Keep the buffer below the electrode plugs.
10. Place a magnetic stir bar in the bottom of the box. Move box to 4� coldroom. Buffer must be stirred during blotting run. Set magnetic stirrer at speed 6.
11. Hoeffer/LKB Transphor 10 v/30 min then 40 v/2 hours or 20 v/overnight
BioRad Transblot 20 v overnight. Do not use the 40 volt protocol with the BioRad unit.
12. UV fix the wet membrane (leave it on the wet 3MM support) with 120,000 �oules UV via the UV crosslinker. Alternatively, use a Sylvania germicidal UV lamp exactly 12 cm from the bulb for exactly 2 min. Caution: nitrocellulose is highly flammable. Air dry the membrane. Place a single membrane in a hybridization bag. Seal the bag. Cut one corner and add prehyb solution at 5 mL/100 cm2 membrane. Incubate in shaking waterbath for 3 hours at 42�. Cut a corner of the bag and remove all prehyb. Add hybridization solution at 5 mL/100 cm2 membrane.
13. Add 1 x 107 cpm 32 P-labeled probe to 300 � reserved hyb mix and boil for 3-5 min. Transfer the probe to ice and then add the probe to the hybridization bag within 20 min. Seal the bag. Rinse the bag in the sink to remove prehyb or hyb mix from the outside of the bag, as these will support the growth of bacteria in the waterbath. Incubate the hyb solution in a shaking waterbath overnight at 55�. Alternatively, a rotisserie apparatus can be used instead of hybridization bags and shaking water baths.
13. Prehybridization mix (add in order listed at 5 mL/100 cm2 membrane)
5x Denhardt�, 5x SSC, 50 mM NaPO4 , pH 6.7, 50% deionized formamide250 �/mL sheared salmon sperm DNA, 1% dextran sulfate MW 5000 and 0.1% SDS
Prep the hyb mix and prehyb mix fresh each time. The hyb mix can sit at room temperature during the 3 hour prehyb step.
14. Hybridization mix (add in order listed at 5 mL/100 cm2 membrane)
14. Hybridization mix (add in order listed at 5 mL/100 cm2 membrane):
1x Denhardt�, 5x SSC, 20 mM NaPO4 , pH 6.7, 50% deionized formamide, 100 �/mL sheared salmon sperm DNA, 5% dextran sulfate MW 5000, and 0.1% SDS
15. Wash square Pyrex dish that will fit in shaking waterbath. Soak in 3% hydrogen peroxide or 0.1% DEPC for 10 min.
16. High Stringency Washes 1, 2 and 3
Wash 1 2x SSC: 80 mL 25x SSC, pH 7.0, 910 mL Type I water, 10 mL 10% SDS
Wash 2 1x SSC: 40 mL 25x SSC, pH 7.0, 940 mL Type I water, 10 mL 10% SDS
Wash 3 0.5x SSC: 20 mL 25x SSC, pH 7.0, 970 mL Type I water, 10 mL 10% SDS
17. Discard the probe and hybridization solution in 32 P liquid waste. Rinse the bag with approximately 30 mL wash 1 and discard the wash in 32 P liquid waste. Discard the 0.1% DEPC in the tray and add approximately 100 mL wash 1 to the dish. Transfer the membrane to the dish and cover with Saran. Shake gently at room temperature for 10 min. Discard wash in 32 P waste. Microwave the remaining wash for 1 min to prewarm slightly and add to the Pyrex dish containing the probed membrane. Incubate at 65� for 30 min. Washes may be performed in the hybridization bag, but may result in higher background. Discard wash 1 and add 1000 mL prewarmed wash 2 and incubate at 65� for 30 min. Discard wash 2 and add 1000 mL prewarmed wash 3 and incubate at 65� for 30 min.
18. Air dry the filter on 3MM paper and wrap with Saran. Expose the membrane as desired.
Do not let the membrane dry out if reprobing is required. Place the blot in a clear plastic sheet protector and seal it closed with tape for chemiluminescent exposure. For storage, seal the blot in a water-tight seal-a-meal pouch and store at ?0�. Note that only SaranWrap is water-tight. Other plastic wraps are water-permeable and allow blots to dry out (and allow water from wet gels to contact autoradiography film, causing black spots).
Neutral membranes have an equal and uniform distribution of charges. Positively charged membranes have a preponderance of positive charges across the matrix. They will hold the same amount of nucleic acid. Positively charged membranes have stronger retention: the target remains bound longer. UV crosslinking forms a covalent bond between nucleic acids and the positive charges of the membrane. Fixation by baking creates a hydrophobic interaction between the nucleic acids and the membrane. If stripping and reprobing is required, it is better to UV crosslink to a positively charged membrane in order to preserve the integrity of the target during the stripping step.
Quality membranes do not have a correct side. The charges are equally distributed on both sides.
Nominally, 0.22 � pore membranes have greater surface area than 0.45 � membranes. They also require better blocking. Consistent pore size is critical. For Northern blots of small RNAs (~ 100 nucleotides), 0.1 � membranes are recommended.
Particulates in unfiltered buffer, skin oils, nonspecific antibody binding, addition of too much probe, inadequate post-hybridization washing, improper wash temperature, and other factors can create background fog or spots on membranes.
Blocking can reduce background. Non-specific proteins block certain electrostatic interactions which may attract proteins randomly. Milk proteins, BSA, or commercial blocking agents are typically used.
1. A Hoeffer/LKB Transphor unit is recommended for Northern blots. The Bio-Rad unit has been known to melt when incorrect voltage was applied.
The recommended transfer membrane for very small RNA was 0.1 � nylon (Schleicher & Schuell Nytran or ICN Biotrans) but this material is no longer available. RNA that is < 110 nucleotides will tend to go through a 0.2 � membrane. Try Schleicher & Schuell nitrocellulose membranes (BA79/0.1 � and BA75/0.05 �)? Nylon (neutral or charged) membranes are rated for denatured > 50 bp DNA fragments, require alkaline transfer conditions and may have higher backgrounds.
Hoeffer sponges are 6?x 9?and the gel must be cut to fit these dimensions.
The Hoeffer gel box can blot 4 gels simultaneously if additional cassettes are purchased.
The Hoeffer unit generates more heat than the BioRad unit.
BioRad sponges are 6 1/8?x 8 3/8?and the gel must be cut to fit these dimensions. The BioRad box is limited to a single gel.
Polyacrylamide/TBE gels ranging from 4 to 8% have been successfully used, with or without urea. It is not necessary to presoak the gel to remove urea prior to blotting. If the gel is severely overloaded, some RNA will transfer to a second layer of nylon.
2. Water and buffers do not need to be DEPC-treated if they do not come from carboys. Carboy spouts tend to be a source of mildew RNases that are resistant to DEPC.
3. Prep a clean tray large enough to hold the blot cassette and soak in 0.1% DEPC or 3% hydrogen peroxide for 10 min at room temperature. Cover the pan during the soak.
4. Prep 5000 mL 1x TAE5 for the Hoeffer unit or 3300 mL 1x TAE5 for the BioRad unit. Pre-chill the buffer on ice in the coldroom.
5. Cut nylon or nitrocellulose and 2 pieces of Whatman 3MM to required dimensions while wearing gloves to prevent finger oils from coming into contact with membrane. Clip one corner to indicate orientation.
6. Drain 0.1% DEPC or 3% hydrogen peroxide from the tray. Rinse with a trace of 1x TAE5 . Add remaining portion of 1 liter 1x TAE5 to tray. Add plastic gel cassette and sponges. Squeeze the air out of the sponges. Soak the membrane briefly by laying it on the sponges.
7. Open the gel plates. Apply membrane to gel and gently brush out air bubbles. Dip one piece of 3MM very briefly in 1x TAE5 and lay on top of membrane. Remove air bubbles. Lay glass plate on 3MM and flip gel over. Remove the other plate (this seems to be difficult) and prewet the remaining piece of 3MM. Lay this on top of the gel. Remove air bubbles. Move to plastic cassette and assemble holder.
8. Place the membrane side of the gel cassette towards the red electrode. Add prechilled 1x TAE5 buffer to cover. Use the 1x TAE5 buffer in the presoak tray as part of the required buffer.
9. Careful! Electrode plugs on the BioRad Transblot will corrode when wet. Keep the buffer below the electrode plugs.
10. Place a magnetic stir bar in the bottom of the box. Move box to 4� coldroom. Buffer must be stirred during blotting run. Set magnetic stirrer at speed 6.
11. Hoeffer/LKB Transphor 10 v/30 min then 40 v/2 hours or 20 v/overnight
BioRad Transblot 20 v overnight. Do not use the 40 volt protocol with the BioRad unit.
12. UV fix the wet membrane (leave it on the wet 3MM support) with 120,000 �oules UV via the UV crosslinker. Alternatively, use a Sylvania germicidal UV lamp exactly 12 cm from the bulb for exactly 2 min. Caution: nitrocellulose is highly flammable. Air dry the membrane. Place a single membrane in a hybridization bag. Seal the bag. Cut one corner and add prehyb solution at 5 mL/100 cm2 membrane. Incubate in shaking waterbath for 3 hours at 42�. Cut a corner of the bag and remove all prehyb. Add hybridization solution at 5 mL/100 cm2 membrane.
13. Add 1 x 107 cpm 32 P-labeled probe to 300 � reserved hyb mix and boil for 3-5 min. Transfer the probe to ice and then add the probe to the hybridization bag within 20 min. Seal the bag. Rinse the bag in the sink to remove prehyb or hyb mix from the outside of the bag, as these will support the growth of bacteria in the waterbath. Incubate the hyb solution in a shaking waterbath overnight at 55�. Alternatively, a rotisserie apparatus can be used instead of hybridization bags and shaking water baths.
13. Prehybridization mix (add in order listed at 5 mL/100 cm2 membrane)
5x Denhardt�, 5x SSC, 50 mM NaPO4 , pH 6.7, 50% deionized formamide250 �/mL sheared salmon sperm DNA, 1% dextran sulfate MW 5000 and 0.1% SDS
Prep the hyb mix and prehyb mix fresh each time. The hyb mix can sit at room temperature during the 3 hour prehyb step.
14. Hybridization mix (add in order listed at 5 mL/100 cm2 membrane)
14. Hybridization mix (add in order listed at 5 mL/100 cm2 membrane):
1x Denhardt�, 5x SSC, 20 mM NaPO4 , pH 6.7, 50% deionized formamide, 100 �/mL sheared salmon sperm DNA, 5% dextran sulfate MW 5000, and 0.1% SDS
15. Wash square Pyrex dish that will fit in shaking waterbath. Soak in 3% hydrogen peroxide or 0.1% DEPC for 10 min.
16. High Stringency Washes 1, 2 and 3
Wash 1 2x SSC: 80 mL 25x SSC, pH 7.0, 910 mL Type I water, 10 mL 10% SDS
Wash 2 1x SSC: 40 mL 25x SSC, pH 7.0, 940 mL Type I water, 10 mL 10% SDS
Wash 3 0.5x SSC: 20 mL 25x SSC, pH 7.0, 970 mL Type I water, 10 mL 10% SDS
17. Discard the probe and hybridization solution in 32 P liquid waste. Rinse the bag with approximately 30 mL wash 1 and discard the wash in 32 P liquid waste. Discard the 0.1% DEPC in the tray and add approximately 100 mL wash 1 to the dish. Transfer the membrane to the dish and cover with Saran. Shake gently at room temperature for 10 min. Discard wash in 32 P waste. Microwave the remaining wash for 1 min to prewarm slightly and add to the Pyrex dish containing the probed membrane. Incubate at 65� for 30 min. Washes may be performed in the hybridization bag, but may result in higher background. Discard wash 1 and add 1000 mL prewarmed wash 2 and incubate at 65� for 30 min. Discard wash 2 and add 1000 mL prewarmed wash 3 and incubate at 65� for 30 min.
18. Air dry the filter on 3MM paper and wrap with Saran. Expose the membrane as desired.
Do not let the membrane dry out if reprobing is required. Place the blot in a clear plastic sheet protector and seal it closed with tape for chemiluminescent exposure. For storage, seal the blot in a water-tight seal-a-meal pouch and store at ?0�. Note that only SaranWrap is water-tight. Other plastic wraps are water-permeable and allow blots to dry out (and allow water from wet gels to contact autoradiography film, causing black spots).
Neutral membranes have an equal and uniform distribution of charges. Positively charged membranes have a preponderance of positive charges across the matrix. They will hold the same amount of nucleic acid. Positively charged membranes have stronger retention: the target remains bound longer. UV crosslinking forms a covalent bond between nucleic acids and the positive charges of the membrane. Fixation by baking creates a hydrophobic interaction between the nucleic acids and the membrane. If stripping and reprobing is required, it is better to UV crosslink to a positively charged membrane in order to preserve the integrity of the target during the stripping step.
Quality membranes do not have a correct side. The charges are equally distributed on both sides.
Nominally, 0.22 � pore membranes have greater surface area than 0.45 � membranes. They also require better blocking. Consistent pore size is critical. For Northern blots of small RNAs (~ 100 nucleotides), 0.1 � membranes are recommended.
Particulates in unfiltered buffer, skin oils, nonspecific antibody binding, addition of too much probe, inadequate post-hybridization washing, improper wash temperature, and other factors can create background fog or spots on membranes.
Blocking can reduce background. Non-specific proteins block certain electrostatic interactions which may attract proteins randomly. Milk proteins, BSA, or commercial blocking agents are typically used.