Two-hybrid analysis of genetic regulatory networks
互联网
<center> <h3> <font><font>Table of contents<br /> </font> </font></h3> </center>
1. Introduction and Background
2. Interaction mating
2.1 Interaction mating - small scale
Protocol 1. Mating assay - small scale for tens of different bait or prey proteins.
2.2 Interaction mating - large scale
Protocol 2. Collecting bait (and prey) strains
Protocol 3. Mating assay - large scale for hundreds of different bait or prey strains.
3. Interaction mating assay with other yeast two-hybrid systems
4. Recording the results
5. Interpreting interaction data
5.1 Qualitative interpretation
5.2 Quantitative interpretation
5.3 Inference of function from pattern of interactions
6. Library scale and genome-wide characterization of protein networks
7. Conclusions
8. Acknowledgments
9. References
1. Introduction and Background
There is a great need for general methods to characterize the proteins that contemporary biology makes available. The list of such proteins needing further characterization is growing and includes proteins already known to be important for specific cellular functions, mutant proteins identified in vivo or made in vitro , and very large numbers of protein being identified by genome projects. Here we describe the extension of two-hybrid approaches so that they can bear on this problem.
The recent success of two-hybrid systems is due to the fact that many cellular functions are carried out by proteins that touch one another. For example, the complex process of transcription initiation requires the ordered assembly of numerous interacting transcription factors with RNA polymerase and ancillary proteins, into a protein machine that initiates transcription (Guarente, 1996; Tjian and Maniatis, 1994). This machine can be viewed as a network of interacting proteins, as can the machines that control other processes, such as DNA replication, protein translation, and the cell cycle. A full understanding of these processes will require knowledge of, not only the proteins (parts) that make up each machine, but also of the topological relationships (connections) that individual parts make with one another.
Likewise, a full understanding of the function of any new protein will require knowledge of the interactions it makes with previously identified proteins. Currently, most new proteins are being identified by large scale sequencing projects. For many of these new proteins the sequence alone sheds little or no light on their function.
Two-hybrid systems have been used to probe the function of new proteins ever since they were developed (Chien et al., 1991; Fields and Song, 1989). The first application of two-hybrid methods to probe protein function was to examine the interactions between proteins isolated by two hybrid methods and relatively small numbers of test proteins (see for example, Durfee et al., 1993; Gyuris et al., 1993; Harper et al., 1993; Zervos et al., 1993), but their use quickly spread to the analysis of many other proteins (Choi et al., 1994; Kranz et al., 1994; Marcus et al., 1994; Printen and Sprague, 1994; Van Aelst et al., 1993; Yuan et al., 1993). In anticipation of the utility of applying these methods to larger sets, we and others began devising ways to do so.
Larger scale two hybrid approaches typically rely on interaction mating. In this method the protein fused to the DNA-binding domain (the bait) and the protein fused to the activation domain (here called the prey) are expressed in two different haploid yeast strains of opposite mating type (MATa and MATa), and the strains are mated to determine if the two proteins interact. Mating occurs when haploid yeast strains of opposite mating type come into contact, and results in fusion of the two haploids to form a diploid yeast strain. Thus, an interaction can be determined by measuring activation of a two-hybrid reporter gene in the diploid strain.
As described below, interaction mating has been used to examine interactions between small sets of tens of proteins (Finley and Brent, 1994; Finley and Brent, 1995; Reymond and Brent, 1995), larger sets of hundreds of proteins (R.L.F. and R.B., unpublished), to screen libraries (Bendixen et al., 1994), and to attempt to comprehensively map connections between proteins encoded by a small genome (Bartel et al., 1996). The primary advantage of this technique is that it reduces the number of yeast transformations needed to test individual interactions. For example, to test for interactions between a set of 10 bait proteins and 5 prey proteins without interaction mating would require 50 transformations to create 50 strains that carry the pair-wise combinations of baits and preys. With mating however, only 15 transformations would be needed; 10 for the different bait plasmids, and 5 for the different prey plasmids; and the resulting two sets of transformants would be mated to create the 50 combinations. The microbiology of the mating procedure (which is extremely simple) is detailed in Section 2.
Interaction mating techniques have facilitated a number of two-hybrid studies of protein protein interaction. Among its first uses was to determine the specificity of interactors isolated in library screens or interactor hunts (Harper et al., 1993). As described in the previous chapters, in the first steps of an interactor hunt, one isolates genes that encode proteins that interact with a particular bait. Before the interacting proteins are further characterized, it is necessary to determine if their interaction with the bait is specific by showing that they do not interact with other unrelated baits or with the DNA-binding domain portion of the bait. When mating is used to test specificity, the strain that contains the activation domain fused protein (prey) is mated with different yeast strains which express either the original bait protein or other, preferably unrelated baits, and the investigator verifies that the reporters are only active in diploids that contain the original bait (Finley and Brent, 1994; Finley and Brent, 1995; Harper et al., 1993).
For example, Harper, Elledge and colleagues used a mating assay to test the specificity of newly isolated interactors (Harper et al., 1993). The methods of these investigators also circumvented the need to isolate the prey plasmid. In their experiments, they performed two-hybrid hunts with a bait plasmid that contains a dominant marker, CYH2 , that can be selected against by plating the yeast on medium containing cycloheximide, which is toxic to yeast that carry CYH2 . Yeast isolated in an interactor hunt were plated on cycloheximide plates to select those that had lost the original bait plasmid but retained the library plasmid. The resulting strain was then mated with a collection of bait strains, including ones that expressed the original bait, to determine the specificity of the library-encoded prey. A mating scheme has also been used directly in an interactor hunt by mating a strain expressing a bait with a strain transformed with the library DNA; here, mating promises to bypass the need to perform separate transformations with library DNA for each new hunt (Bendixen et al., 1994).
In addition to its use in interactor hunts, mating can be used to characterize small sets of proteins as described in Section 2.1 and Protocol 1. In one example of this approach, we used interaction mating to characterize a set of seven Drosophila Cyclin-dependent kinases (Cdk) interactors, or Cdis (Finley and Brent, 1994). Strains expressing versions of the Cdis fused to an activation domain were mated with 74 different strains expressing different bait proteins, including Cdks from other species and four of the Cdis themselves. The results from this study illustrate the types of information that can be derived from such a characterization. First, the experiments showed that some of the Cdis interacted with different subgroups of seven highly related Cdk baits, suggesting that the Cdis recognize structural features shared by these Cdks but absent in the non-interacting Cdks; inspection of an alignment of the Cdk protein sequences suggested residues that may be important for specific interactions with certain Cdis. Second, Cdi3, Drosophila Cyclin D, interacted much more strongly with human Cdk4 than with any of the other Cdks in the panel including the Drosophila Cdks, suggesting that there may be an as yet unidentified Drosophila Cdk4 homolog which is the true partner for Cyclin D. Third, two of the Cdis interacted with two other Cdis, indicating in each instance that each Cdi has surfaces for binding to the Cdk and to another Cdi, and suggesting that these proteins form ternary or higher order complexes. Finally, the demonstration that two Cdis with no sequence similarity to previously identified proteins interact with each other as well as with the Cdk, but not with a panel of over 60 other proteins, provided an additional clue to their functions, strongly supporting the idea that they function along with the Cdk in the network of proteins that regulates the cell cycle. These results demonstrate that examination of the interactions between even small numbers of proteins can provide a number of functional insights. Much larger sets of proteins can be characterized by scaling up these procedures as described in Section 2.2 and discussed in Sections 6 and 7.
2. Interaction mating
In this section we present methods for performing interaction mating assays on small or large sets of proteins using the interaction trap, and in Section 3 we discuss use of interaction mating with other two-hybrid systems. The interaction trap (see Chapter 4 and references therein) uses the E.coli protein LexA as the DNA-binding domain and a protein encoded by random E. coli sequences, the B42 "acid blob", as the transcription activation domain. Both proteins are expressed from multicopy (2µ) plasmids; the LexA fusion, or bait, is expressed from a plasmid containing the HIS3 marker, and the activation domain fused protein, or prey, is expressed from a plasmid containing the TRP1 marker. In the most commonly used bait plasmid, pEG202, the bait is expressed from the constitutive yeast ADH1 promoter. Related bait plasmids are available which express the bait fused to a nuclear localization signal (pNLex, see Chapter 4), or which express the bait conditionally from the GAL1 promoter (pGILDA, D. Shaywitz and C. Kaiser, personal communication). The most commonly used prey plasmid, pJG4-5, expresses proteins fused to the B42 activation domain, the SV40 nuclear localization signal, and an epitope tag derived from hemagglutinin, all driven by the yeast GAL1 promoter which is active only in yeast grown on galactose (Gyuris et al., 1993). Use of the GAL1 promoter to express the prey allows toxic proteins to be expressed transiently and helps eliminate many false positives in interactor hunts (Chapter 4). The interaction trap uses two reporter genes that carry upstream LexA binding sites (operators): LEU2 and lacZ . The LEU2 reporters are integrated into the yeast genome and the lacZ reporters typically reside on 2µ plasmids bearing the URA3 marker, though integrated versions are also available (R.L.F., R.B., S. Hanes, unpublished). Several versions of the LEU2 and lacZ reporters have been made that have a range of sensitivities based on the number of upstream LexA operators. In general the LEU2 reporters are more sensitive to a given interacting pair of proteins than the lacZ reporters (Estojak et al., 1995); however, recently highly sensitive lacZ reporters have been used that contain several LexA operators and transcription terminator sequences downstream of the lacZ gene (S. Hanes, personal communication).
Several different combinations of strains, plasmids, and reporters can be used for mating (Section 3). In one common version (Finley and Brent, 1994), the strain expressing the bait (bait strain) is RFY206 (MATa ura3-52 his3Æ200 leu2-3 lys2Æ201 trp1::hisG ) transformed with the HIS3 bait plasmid and a URA3 lacZ reporter plasmid like pSH18-34. The strain expressing the activation domain-tagged protein (prey strain) is EGY48 (MATa ura3 his3 leu2::3LexAop-LEU2 trp1 LYS2 ) transformed with the TRP1 prey plasmid. Patches of these two strains on agar plates are brought into contact by replica plating (see below) and grown on a rich medium overnight. During this time cells in the patches mate and fuse to form diploids. The cells are then transferred by replica plating to plates on which only diploids can grow: these plates lack uracil, histidine, and tryptophan so that neither parental haploid can grow on them. To avoid an additional step, the diploid selection plates are also indicator plates, which allows an interaction to be scored by testing for expression of the reporter genes. In the protocols presented here the lacZ reporter is measured, using diploid selection indicator plates containing X-Gal, a chromogenic substrate for the lacZ gene product. However, it is worth mentioning that expression of the LEU2 reporter can also be easily scored by putting the diploids on plates that lack leucine, and that the future will likely bring other reporters. Furthermore, because both reporter genes exhibit a reduced sensitivity in diploid strains compared to haploid strains, the most sensitive versions of the lacZ or LEU2 reporters are recommended for interaction mating assays.
Variants of this simple procedure are sometimes useful. In particular, because some baits activate transcription by themselves, it is often useful to conditionally express the prey protein so that one scores patches that show an increase in reporter gene expression in the presence of the prey. To do this, the diploids are placed on two different X-Gal plates, one that contains galactose, which results in expression of the prey, and one that contains glucose which represses expression of the prey. Here, an interaction between the bait and prey is detected when the diploid yeast containing them turn more blue on the galactose X-Gal plate than on the glucose X-Gal plate.
2.1 Interaction mating - small scale
It is often informative to look for interactions between small sets of proteins, or between a given protein and a test set of ten to a hundred proteins. The test set, for example, might contain different allelic forms of the original bait, sets of structurally related proteins, sets of proteins known or suspected to be involved in some process, and unrelated proteins used to demonstrate the specificity of an interaction. Protocol 1 describes a convenient method to test small sets of proteins for interactions.
The collections of bait and prey strains used here can be maintained on yeast plates stored at 4oC for two to three months, or stored frozen for several years (see Protocol 2). For mating, the two strains are first streaked to the appropriate selection plates: the bait strains (RFY206 containing the URA3 lacZ reporter plasmid and HIS3 bait plasmid) are streaked to plates lacking uracil and histidine -u-h Glu) to maintain selection for the two plasmids; the prey strains (EGY48 containing the TRP1 prey plasmid) are streaked to plates lacking tryptophan (-w Glu) to maintain selection for the prey plasmid. The haploid strains are then brought into contact by placing both plates sequentially on the same replica velvet and lifting the double imprint with a YPD plate (see Protocol 1). If the bait strains are streaked in parallel horizontal stripes and the prey strains are streaked in vertical stripes, physical contact between the strains will occur at the intersections of the stripes on the YPD plate. After a brief period of growth to allow diploids to form, the yeast are transferred to diploid selection indicator plates by replica plating. Diploid colonies that contain a pair of interacting bait and prey proteins are more blue on the galactose X-Gal plate than the glucose X-Gal plate.
________________________________________________________________________
Protocol 1. Mating assay - small scale for tens of different bait or prey proteins.
Materials
_ Bait strains: S. cerevisiae strain RFY206 (MATa ura3-52 his3Æ200 leu2-3 lys2Æ201 trp1::hisG ) transformed with a URA3 plasmid containing a lacZ reporter, such as pSH18-34, and various HIS3 bait plasmids, such as derivatives of pEG202 that produce different LexA fusions. Each bait strain will contain a different bait plasmid.
-
Prey strains: S. cerevisiae strain EGY48 (MATa ura3 his3 leu2 ::3LexAop-LEU2 trp1 LYS2) transformed with TRP1 prey plasmids, such as derivatives of pJG4-5 that produce different activation domain-tagged proteins or preys
-
Sterile wooden applicator sticks (e.g. FisherBrand 01-340)
-
Minimal glucose yeast plates lacking uracil and histidine (-u-h Glu), see Chapter 4
-
Minimal glucose plates lacking tryptophan (-w Glu), see Chapter 4
-
YPD plates, see Chapter 4
-
Minimal X-Gal glucose plates lacking uracil, histidine, and tryptophan (-u-h-w Glu X-Gal), see Chapter 4.
-
Minimal X-Gal galactose/raffinose plates lacking uracil, histidine, and tryptophan (-u-h-w Gal/Raf X-Gal) , see Chapter 4
-
Replica plater and sterile replica velvets
Optional
-
Minimal glucose plates lacking uracil, histidine, tryptophan, and leucine (-u-h-w-l Glu), see Chapter 4
-
Minimal galactose/raffinose plates lacking uracil, histidine, tryptophan, and leucine (-u-h-w- Gal/Raf), see Chapter 4
Method
1. Streak different bait strains in horizontal parallel stripes on a -u-h Glu plate. Streaks should be at least 3 mm wide and at least 5 mm apart, with the first streak starting about 15 mm from the edge of the plate. A 100 mm plate (which for some reason is typically 90 mm in diameter) will hold 8 different bait strains. Create a duplicate plate of bait strains for each different plate of prey strains to be used.
2. Likewise, streak different prey strains in vertical parallel stripes on a -w Glu plate. As a control for baits that may activate transcription, include a prey strain that contains the prey vector pJG4-5 not encoding a fusion protein (i.e. encoding only the activation domain). Create a duplicate plate of prey strains for each plate of bait strains to be used.
3. Incubate plates at 30oC until there is heavy growth on the streaks. When taken from reasonably fresh cultures, for example plates that have been stored at 4oC for less than a month, streaked RFY206-derived bait strains take about 48 hours to grow and EGY48-derived prey strains take about 24 hours.
4. Press a plate of prey strains to a replica velvet, evenly and firmly so that yeast from all along each streak are left on the velvet. This plate may be reused if necessary. Press a plate of bait strains to the same replica velvet. This plate of bait strains cannot be reused as it is now contaminated with prey strains.
5. Lift the impression of the bait and prey strains from the velvet by pressing a YPD plate on it. Incubate the YPD plate for 24 hours at 30oC.
6. Replica YPD plates to the following diploid selection, indicator plates: -u-h-w Glu X-Gal, -u-h-w Gal/Raf, and (optional: -u-h-w-l Glu, and -u-h-w-l Gal/Raf). The YPD plate should contain sufficient growth to enable a single impression on the velvet to be lifted by at least four indicator plates.
7. Patch control strains (see text) onto the indicator plates and incubate at 30oC. Examine results daily. Diploids will grow and blue color will develop within 2 days.
________________________________________________________________________
2.2 Interaction mating - large scale
With a few modifications, the procedure described above can be used to test for interactions between a single prey protein and hundreds of baits (Protocol 3, see Figure 1 below). Large panels of bait strains can be collected and stored frozen indefinitely (Protocol 2) and then screened against any number of preys. One such set of bait strains contains over 700 different LexA fusion proteins from our own work and from numerous other labs that use the interaction trap (R.L.F., R.B., A. Reymond, unpublished). Screening a protein against such a panel enables one to quickly test its ability to interact with a large number of known proteins, most of which have been characterized to some extent, and have been chosen for study because of their known or suspected involvement in some biological process. Thus, the finding of an interaction between a tested protein and a member of the panel can often lead to immediate clues about the biological function of both proteins (see Section 5). While the number of proteins in the existing panel is far less than the number of proteins in a good library, this approach does offer the advantage of screening the test protein against a set of proteins enriched for those of current interest to the biological community. It is worth noting that these proteins come from many different organisms in which they are expressed in different tissues and at different developmental stages. Thus it becomes possible to identify interacting partners that have not yet been isolated from the same species, or that are not expressed in tissues from which interaction libraries have been made.
For some proteins, this approach offers additional advantages over screening a library using a traditional two-hybrid scheme. Proteins that activate transcription when fused to LexA or another DNA-binding domain can be difficult to use in conventional interactor hunts. Though methods are available to reduce the sensitivity of the reporter genes (Durfee et al., 1993; Estojak et al., 1995; Chapter 2, 3, 4) it is not always possible to reduce the reporter sensitivity below the threshold of activation for some baits. Moreover, reduction in reporter sensitivity carries with it the risk that the reporters will not detect weakly interacting proteins. Furthermore, spontaneously occurring yeast mutations, for example those that increase the copy number of the bait plasmid, can increase the activating potential of weakly activating baits (R.L.F., R.B., A. Mendelsohn, unpublished data); such mutations are typically scored as positive in the early stages of an interactor hunt, and they are not readily detected in schemes where the specificity test is performed by removing the bait plasmid from the strain containing the prey and mating the strain with other bait strains. Thus, an alternative for proteins that activate transcription as baits, is to use them as preys to screen existing panels of baits, or even libraries of baits. Interaction mating approaches also have clear advantages for proteins that are somewhat toxic to yeast; the prey vector allows conditional expression of toxic proteins in the presence of a bait, and often the interaction can be observed as the reporters are activated even if the cells are inviable. An example of the use of interaction mating together with a large panel of bait strains to characterize a protein that both activates transcription and is toxic to yeast, Drosophila Cyclin E (Finley, Zavitz, Thomas, Richardson, Zipursky, and Brent, in prep), is discussed in Section 7.
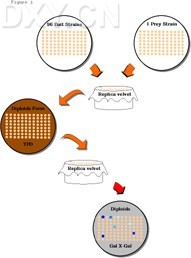
Figure 1. Mating assay for interactions between a prey and 96 baits
Figure 1.
Top. The plate on the left holds 96 different yeast strains in patches (or colonies) that each express a different bait protein. The plate on the right holds 96 patches, each of the same yeast strain (prey strain) that expresses a protein fused to an activation domain (prey). The plate of bait strains and the plate of prey strains are each pressed to the same replica velvet and the impression is lifted with a plate containing YPD medium. After one day of growth on the YPD plate, during which time the two strains mate to form diploids, the YPD plate is pressed to a new replica velvet and the impression is lifted with a plate containing diploid selection medium and an indicator like X-Gal. Blue patches (dark spots) on the X-Gal plate indicate that the lacZ reporter is transcribed, suggesting that the prey interacts with the bait at that location.
________________________________________________________________________
Protocol 2. Collecting bait (and prey) strains
Materials
-
Freezing media: 1:1 solution of minimal glucose media lacking appropriate amino acids (e.g. -u-h Glu for bait strains) : sterile glycerol solution (65% (v/v) glycerol, 0.1 M MgSO4, 25 mM Tris-HCl pH 7.4)
-
1.0 to 1.5 ml cryotubes
-
Yeast strains freshly streaked to minimal glucose plates
-
Sterile wooden applicator strips
Methods
1. Streak bait strains to -u-h Glu plates, or prey strains to -w Glu plates, and incubate at 30oC for 24 to 48 hours. Yeast should be taken from the plates and frozen no more than 4 days after being streaked.
2. With a sterile wooden applicator stick, grab a dollop of yeast from the plates and inoculate 0.5 ml of freezing solution in a cryotube. Vortex lightly. This solution should have an OD600 over 3.0.
3. Alternatively, inoculate 0.5 ml of -u-h Glu liquid media to an OD600 less than 0.2, incubate at 30oC with shaking until OD600 = 1.5 to 2.0 (log phase), and add 0.25 ml of this culture to 0.25 ml of sterile glycerol solution in a cryotube.
4. Freeze by placing cryotubes in -80oC freezer. Most strains can be recovered after up to at least two years by scraping the surface of the ice and streaking to minimal glucose plates. Avoid allowing entire contents of cryotube to thaw.
________________________________________________________________________
________________________________________________________________________
Protocol 3. Mating assay - large scale for hundreds of different bait or prey strains.
Materials
-
Freshly streaked bait and prey strains (see Protocol 1)
-
One set of the following 150 x 15 mm plates for each test of interactions between an activation domain-tagged protein (in a prey strain) and 96 baits (bait strains): -u-h- Glu; -w Glu; YPD; -u-h-w Glu X-Gal; -u-h-w Gal/Raf X-Gal
-
Replica plater and sterile velvets for 150 mm diameter plates. (A replica devise can be fashioned from a box of 200 µl pipet tips by stretching a velvet over the top of the box)
-
96-prong device (e.g. DanKar MC-96) with 3 mm diameter flat ended metal prongs in a 96-well configuration. Similar devices can be used in 48-well configurations for use with 100 mm plates.
-
0.5 to 4.0 ml sterilized tubes arranged in a 96-well configurations (e.g. cluster tubes such as Costar #4411). Ideally these tube can be capped and frozen at -80oC.
-
-u-h Glu liquid media, see Chapter 4
-
-w Glu liquid media, see Chapter 4
-
Sterile glycerol solution (65% (v/v) glycerol, 0.1 M MgSO4, 25 mM Tris-HCl pH 7.4)
Methods
1. It is most convenient to place large numbers of bait strains in a 96-well configuration ( Figure 1 ). This can be done by inoculating 2 ml of -u-h Glu media in cluster tubes and growing to OD600 = 1.5 to 2.0. After making plates from these cultures (see step 2 below) add an equal volume of sterile glycerol solution, cap and freeze at -80oC.
2. Use the 96-prong device, sterilized in ethanol and flame, to transfer bait strains from the culture to the center of a 150 mm -u-h Glu plate. Each plate can contain 96 different bait strains. Tens of identical plates can be made from one culture. Incubate the plates at 30oC for 48 hours or until all bait strains have grown to colonies 5 mm in diameter. These plates can be stored at 4oC for up to 2 months and used to inoculate another liquid culture when more plates are needed. Several positions on each plate should contain control strains with baits that activate various levels of transcription (see Section 4 and Table 1 ).
3. Inoculate 50 ml of -w Glu liquid media with a prey strain and grow at 30oC with shaking to OD600 = 1.5 to 3.0. Pour the culture into a sterile 150 mm plate, or into the sterile top from a box of 200 µl pipets, and use the 96-prong device, sterilized in ethanol and flame, to transfer the culture to -w Glu plates. On these plates, all 96 positions will contain the same prey strain.
4. Follow the replica plating procedure from Protocol 1 to combine the bait and prey strains to a YPD plate, and then after growth on the YPD plate at 30oC for 24 hours, replica to X-Gal indicator, diploid selection plates (-u-h-w Glu X-Gal and -u-h-w Gal/Raf X-Gal) (see Figure 1 above).
5. Examine results after two days.
________________________________________________________________________
3. Interaction mating assay with other yeast two-hybrid systems
In addition to the interaction trap, many other yeast two-hybrid systems have been developed (see Chapter 1 and Allen et al., 1995; Fields and Sternglanz, 1994; Mendelsohn and Brent, 1994, for reviews). All of these allow the analysis of individual protein-protein interactions, and permit interactor hunts to isolate new proteins that interact with a bait. In some instances plasmids or strains from one system can be used in another, but often the components are incompatible. Most often, the yeast selectable markers on the different components differ. In addition, systems that use Gal4 as the DNA binding domain cannot be used with yeast strains that have a wild-type GAL4 gene, and therefore, since the Gal4 protein is required to activate the GAL1 promoter, cannot be used with systems that use the GAL1 promoter to drive expression of the prey protein. Finally, use of interaction mating requires careful attention to the mating types of the strains and the selectable markers used to select the diploids.
4. Recording the results
Interaction between bait and prey results in the interaction phenotypes : growth of the strain on medium lacking leucine, and transcriptional activation of the lacZ reporter and production of active ß-galactosidase. On X-Gal plates the ß-galactosidase cleaves the X-Gal substrate, producing a product which turns the yeast colony blue. The amount of color provides a fast and simple method to approximate the level of lacZ expression in a strain. An interaction is scored when a the diploid colony is more blue on the X-Gal plate containing galactose than the X-Gal plate containing glucose.
Scoring these interactions benefits from inclusion of a number of controls. To control for common variations between the X-Gal plates, it is useful to include control strains that contain baits which activate transcription to varying extents. Table 1 shows some baits with known activating abilities. Inclusion of such strains on every X-Gal plate enables one to normalize the amount of blue produced by an interaction. It is also useful to include a control strain to check that the plates contain the correct carbon sources, and ensure that the GAL1 promoter which drives the expression of the prey protein is activated on the Gal/Raf plates and not the Glu plates. An ideal control of this nature consists of a diploid strain derived from a mating assay, which expresses an interacting pair of bait and prey proteins, such as any one of a number of well-characterized interacting pairs (Finley and Brent, 1994; Gyuris et al., 1993; Zervos et al., 1993). An alternative to using X-Gal plates is to perform a filter lift assay for ß-galactosidase activity in grown diploid colonies (Chapter ). Finally, every bait should be tested to see if, and how much, it activates transcription in the absence of a prey, which can be simply accomplished by mating the bait strains to a strain containing the empty prey vector. Thus, a true interaction with a prey protein is scored when the amount of galactose-dependent activation of the lacZ reporter (e.g. amount of blue) exceeds the amount produced in the absence of a prey.
Table 1. Activating and non-activating baits
5. Interpreting interaction data
5.1 Qualitative interpretation
For large amounts of information flowing from interaction mating experiments, the problem of determining whether individual interactions are meaningful is multiplied. We consider a number of these separately.
True and false positives . Any given interaction with affinity tighter than 10-6 will get detected. Although there may exist a weak positive correlation between apparent tightness and biological significance, many apparently weak interactions are real while some strong ones are not. The problem of determining which interactions have biological significance is therefore not trivial. At the moment, the most satisfying way to show biological significance is to verify the interaction by a different, biochemical technique, preferably co-precipitation from a cell in which both proteins are expressed. However, the interaction data alone can often point out probable true and false positives. For example, our experience indicates that highly specific interactions, such as between a protein that binds to one or a small set of highly related proteins and not to hundreds of unrelated proteins, are good candidates to pursue as biologically relevant. Conversely, we tend to give less weight to interactions between proteins that are sticky, or involving those proteins so ubiquitous in the life of the cell (e.g., members of the ubiquitin system or heat shock proteins) that the interactions might be meaningful but relatively uninformative.
True and false negatives. A problem less frequently considered is that of interactions that are not observed. Two observations suggest that many interactions that should be observed are not. One is that in library screens proteins that should be found occasionally are not. Although failure to recover expected proteins in this instance might be due to trivial considerations, such as the absence of the protein from the library used, another fact suggests there could be other reasons. There are now a number of examples in which known interactions are either not observed, or are subject to directionality, being observed only when one of the two proteins is a bait and the other a prey (see for example, Estojak et al., 1995). Our current doctrine for determining that individual interactions do not occur is that full length and truncated putative partners must be tested in all combinations of baits and preys, with the most sensitive reporters, before the investigator can tentatively conclude that the two proteins do not touch. Since this is impractical for mating experiments that involve a large number of baits and preys, such as the genome wide approaches discussed below, we are resigned that false negatives will arise, and we do not give the absence of interaction any weight in our data analysis. This doctrine may change as more sensitive detection methods are designed.
Multimeric complexes . Finally, it is worth noting that one can build up chains of individual binary interactions to suggest higher order complexes. This has worked well, for example with proteins in signal transduction (Choi et al., 1994; Marcus et al., 1994; Printen and Sprague, 1994), and the advent of mating techniques has made it even easier to build up such patterns (Finley and Brent, 1994; C. Kaiser and D. Shaywitz, personal communication).
5.2 Quantitative interpretation
No two-hybrid technique - particularly the mating techniques described in Protocols 1 and 3 - allows precise quantitation, and any interactions identified must be studied further to determine biological significance and biophysical characteristics. However, some quantitative information does inhere in the data. The amount of ß-galactosidase activity in the cell is proportional to the level of lacZ transcription so that some information about the strength of interaction of two proteins might be derived from measuring ß-galactosidase activity. Though measurement of ß-galactosidase activity with a liquid assay (Guarente and Ptashne, 1981; Rose and Botstein, 1983) is not practical for large numbers of strains, a less precise indication of enzyme activity can be derived from the color of the yeast colony on an X-Gal indicator plate; for example, dark blue, light blue, or white colonies correspond to high, moderate, or low to no lacZ transcription. Despite this correlation between transcription levels and ß-galactosidase activity, one must use caution in using ß-galactosidase activity to compare relative affinities of different bait and prey pairs. Many variables could affect the interaction phenotypes, including the stability of the two fusion proteins, transport of the fusions into the nucleus, and the ability of the bait to bind DNA. These considerations make it imprudent to use two-hybrid data to compare affinities between sets of unrelated proteins.
It is, however, often possible to make meaningful comparisons of the affinity of a single prey protein for several related baits. Such a comparison relies on two assumptions that are generally correct and can be experimentally verified: that the prey, which can be detected with antibodies to its epitope tag, is expressed at the same level in each diploid, and that the baits, which can be detected with anti-LexA antibody and whose DNA binding can be quantitated by a repression assay (Brent and Ptashne, 1984), occupy the operators to similar extents.
5.3 Inference of function from pattern of interactions
One reason for developing interaction mating techniques was the hope that it would reveal contacts between test proteins and known proteins that would provide clues to the function of the test proteins. This turned out be true (see for example, Section 7). However, our first experiments revealed that clues to function might also be derived from the pattern of interactions a protein makes, without reference to the biochemical identity of the interacting proteins. A simple example, taken from our first experiments, illustrates this point. Cdi4 and Cdi11 both interact with Drosophila Cdc2c and interaction mating experiments also revealed that Cdi4 interacts with Cdi11 (Finley and Brent, 1994). From the pattern of interactions alone, these data are consistent with the idea that Cdi4, Cdi11 and Cdc2c could form a three protein complex. It is possible that other such patterns of interactions, particularly conjoined with the crude affinity data, might signal other sorts of regulators. The algorithmic analysis of connectivity data for patterns of this type is an important area of future research.
6. Library scale and genome-wide characterization of protein networks
Interaction mating schemes can also be used on a larger scale, for screening libraries, and, eventually to characterize complex genomes. One such scheme is to mate a pool of cells containing different activation domain-tagged proteins against a bait protein. Another is the converse of the original two-hybrid system. In this approach, a library of different proteins fused to a DNA-binding domain is used in an interactor hunt to find proteins that interact with a specific activation-tagged protein. Historically, the drawback to such approaches has been that libraries that express proteins fused to DNA-binding domains will contain a large number proteins that activate transcription when brought to DNA (Ma and Ptashne, 1987), complicating the task of identifying yeast in which the reporters are active due to the presence of an interacting protein. One way to circumvent this difficulty would be to introduce the library into a yeast strain that contained a counter-selectable reporter gene (e.g. LexAop-LYS2 and LexAop-URA3 ), select against those yeast that contained activators, and then mate the "depleted" library with yeast of the opposite mating type that contain the test protein. Yet another way is to express the activation domain-tagged proteins from a conditional promoter like GAL1 and compare reporter activation between replica plates on which they are and are not expressed, as descried in Protocol 1 and 3, and in Chapter 4).
Recently, Bartel et al applied two-hybrid technology to characterize a small genome (Bartel et al., 1996). They set out to identify all detectable binary interactions between proteins encoded by the bacteriophage T7 genome. They did this by making two libraries, one of DNA-binding domain hybrids and one of activation domain hybrids, expressed in yeast strains of opposite mating type. They then mated a pool of yeast that contained the entire library of activation domain hybrids with 30,000 of the strains expressing DNA-binding domain fusions, in groups of ten so they could readily single out those that activated transcription. They selected diploids in which the HIS3 reporter was activated and screened for activation of a second lacZ reporter using a filter assay. In this way they identified 19 binary interactions between T7 encoded proteins. They further performed individual interactor hunts testing 34 specific DNA-binding hybrids against the entire activation domain library, and 11 specific activation domain hybrids against the entire DNA-binding domain hybrid library, again by interaction mating, and identified 3 additional interactions. Finally, they made a matrix of all of the yeast expressing DNA-binding domain hybrids involved in an interaction mated with yeast expressing all of the activation domain hybrids involved in an interaction to identify three more interactions.
By this means they detected a total of 25 interactions. Some of the interactions were previously known, while others confirmed interactions that had been suspected based on genetic or biochemical studies. Most importantly, 10 of the interactions detected in this two-hybrid tour de force identified connections between proteins not previously known to interact. This new information contains both clues to the function of individual proteins and clues as to how some may function together. An additional windfall from this approach, made possible by the fact that the two libraries were made from random fragments of the T7 genome, was the identification of a number of previously unsuspected intramolecular interactions. The detection of these intramolecular interactions suggested possible homo-oligomeric protein contacts as well as interdomain contacts that might promote the formation of tertiary structure. The success of this genome-wide approach demonstrates that interaction mating techniques can be used to identify the networks of interacting proteins encoded by more complex genomes. The charting of such connections between proteins will provide insights into the functions of individual proteins and lead to a better understanding of how groups of proteins control biological processes.
7. Conclusions
The few years since the advent of two-hybrid systems has proven their utility in the study of defined protein interactions, in identification of new interacting proteins, and in the charting of genetic networks of proteins involved in processes from signal transduction to transcription regulation. These tremendous successes suggest that two-hybrid approaches like those discussed in this chapter may eventually be used to identify all of the protein protein contacts made in a cell or an organism.
Before this time, another need is clear. Sequencing projects like the human genome initiative will soon provide us with the sequences of all of the expressed proteins. A good deal of insight into the function of these proteins can be derived from their sequences alone, but ultimately must be combined with other forms of information to understand the biology in detail. Information about contacts made by the proteins of a genome will complement and augment the sequence information. Such information will likely come from incremental scaling up of the methods described here, as well as from scaled up versions of ideas such as those developed by Bartel et al (Bartel et al., 1996). Connection data will also come from the thousands of labs using two-hybrid systems to identify and characterize specific proteins. Finally, it may also come from recent efforts to identify all of the proteins in the networks of interacting proteins in a cell using rapid sequential two-hybrid interactor hunts that use the proteins isolated in one hunt as starting points for further hunts, in a sort of "protein interaction walk" (R.L.F., unpublished).
As discussed in Section 5, all two-hybrid approaches inevitably produce false positives, interactions that do not occur in any biological setting. Thus, although it will be rich in information, connectivity maps derived from two-hybrid data will necessarily be imprecise. This need not be thought of as a significant drawback of genome-wide two-hybrid approaches, provided it is borne in mind that the information in a protein linkage map derives its utility in providing clues to important interactions which must be explored with further study using other methods.
One example of an insight into protein function from a large scale two-hybrid approach is the identification of the Drosophila protein Roughex, Rux, as a protein that interacts strongly and specifically with Drosophila Cyclin E (Finley, Zavitz, Thomas, Richardson, Zipursky, and Brent, in prep). Rux, a 335 amino acid protein whose sequence gives no clues to its function (Thomas et al., 1994), was in a panel of 600 bait proteins that we tested for interaction with a Cyclin E prey. It was known that rux is required for normal eye development; loss of function rux mutants have rough eyes and aberrant cell cycle regulation in the eye imaginal disc from which the eye develops (Thomas et al., 1994). Thomas et al showed that a stripe of cells in the morphogenetic furrow of the developing eye disc must arrest transiently in the G1 phase of the cell cycle for proper development and this G1 arrest fails in rux mutant eye discs. Combined with this information, the finding that Rux interacts directly with Cyclin E, a protein known to be required for progression through G1, immediately suggested that Rux modulated cyclin activity, and inspired us to undertake specific genetic and biochemical experiments to test the hypothesis.
Scaled up interaction mating assays are likely to be useful in the analysis of genetic diseases and other complex genetic traits. The first version of this idea, which has a long history, is that genes that modify the function of other genes may participate in the same process. A less obvious corollary of this idea became apparent several years ago: that, among the proteins that interact with a protein involved in a disease, those that interact differently with wild-type and disease state allelic forms of the protein are likely to be involved in the disease. Recently, Reymond and Brent undertook a test of this idea (Reymond and Brent, 1995). They studied the protein encoded by the INK4 human tumor suppressor gene, p16. Wild type p16 interacts with two human Cyclin-dependent kinases, Cdk4 and Cdk6 to inhibit their activity. As expected, interaction mating showed that alleles of p16 found in cancer-prone families are deficient in their interaction with the kinases. Two unexpected conclusions arose from these experiments. One allele, p16-G101W, showed decreased interaction with Cdk4 but not with Cdk6, suggesting that its role in disease is unrelated to its action on Cdk6. Furthermore, another allele, p16-I49T, which is also found in the control population, is deficient in interaction with Cdk4, suggesting that this allele may also contribute to a tumor-prone phenotype. These findings underscore the fact that interaction mating with different alleles in a population will contribute to the analysis of complex polygenic traits.
The ability to conduct scaled-up two hybrid analysis has come at a good time. The trickle of new genes and alleles has become a torrent. Robust and general approaches to the understanding of gene and pathway function will help us to the next step of biological understanding.
7. Acknowledgments
We thank L. Lok and members of the Brent laboratory, past and present, for helpful discussions, A. Mendelsohn for assistance in working out the interaction mating assay, and A. Reymond for help in collecting and maintaining the bait panel. We also thank P. Colas, E. Golemis, and C. Giroux for helpful comments on the manuscript. R.B. was supported by Hoescht AG and an American Cancer Society Faculty Research Award.
8. References
Allen, J. B., Walberg, M. W., Edwards, M. C., and Elledge, S. J. (1995). Finding prospective partners in the library: the two-hybrid system and phage display find a match. Trends in Biochem. 20 , 511-516.
Bartel, P. L., Roecklein, J. A., SenGupta, D., and Fields, S. (1996). A protein linkage map of Escherichia coli bacteriophage T7. Nature Genetics 12 , 72-77.
Bendixen, C., Gangloff, S., and Rothstein, R. (1994). A yeast mating-selection scheme for detection of protein-protein interactions. Nucleic Acids Research 22 , 1778-1779.
Brent, R., and Ptashne, M. (1984). A bacterial repressor protein or a yeast transcriptional terminator can block upstream activation of a yeast gene. Nature 312 , 612-615.
Chien, C.-T., Bartel, P. L., Sternglanz, R., and Fields, S. (1991). The two-hybrid system: A method to identify and clone genes for proteins that interact with a protein of interest. Proc. Natl. Acad. Sci. USA 88 , 9578-9582.
Choi, K. Y., Satterberg, B., Lyons, D. M., and Elion, E. A. (1994). Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S. cerevisiae. Cell 78 , 499-512.
Durfee, T., Becherer, K., Chen, P.-L., Yeh, S.-H., Yang, Y., Kilburn, A. E., Lee, W.-H., and Elledge, S. J. (1993). The retinoblastoma protein associates with the protein phophatase type 1 catalytic subunit. Genes and Dev. 7 , 555-569.
Estojak, J., Brent, R., and Golemis, E. A. (1995). Correlation of two-hybrid affinity data with in vitro measurements. Mol Cell Biol 15 , 5820-5829.
Fields, S., and Song, O. (1989). A novel genetic system to detect protein-protein interactions. Nature 340 , 245-246.
Fields, S., and Sternglanz, R. (1994). The two-hybrid system: an assay for protein-protein interactions. Trends Genet. 10 , 286-292.
Finley, R. L., Jr., and Brent, R. (1994). Interaction mating reveals binary and ternary connections between Drosophila cell cycle regulators. Proc Natl Acad Sci U S A 91 , 12980-12984.
Finley, R. L., Jr., and Brent, R. (1995). Interaction trap cloning with yeast. In DNA Cloning 2, Expression Systems: A Practical Approach, B. D. Hames and D. M. Glover, eds. (Oxford: Oxford University Press), pp. 169-203.
Golemis, E.A., and Brent, R. (1992). Fused protein domains inhibit DNA binding by LexA. Mol. Cell. Biol. 12 , 3006-3014.
Guarente, L. (1996). Transcriptional coactivators in yeast and beyond. Trends in Biochem 20 , 517-521.
Guarente, L., and Ptashne, M. (1981). Fusion of Eschericia coli lacZ to the cytochrome c gene of Saccharomyces cerevisiae . Proc. Natl. Acad. Sci. USA 78 , 2199-2203.
Gyuris, J., Golemis, E., Chertkov, H., and Brent, R. (1993). Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell 75 , 791-803.
Harper, J. W., Adami, G. R., Wei, N., Keyomarsi, K., and Elledge, S. J. (1993). The p21 Cdk-interacting protein Cip1 is a potent inhibitor of g1 cyclin-dependent kinases. Cell 75 , 805-816.
Kranz, J. E., Satterberg, B., and Elion, E. A. (1994). The MAP kinase Fus3 associates with and phosphorylates the upstream signaling component Ste5. Genes Dev 8 , 313-27.
Lech, K., Anderson, K., and Brent, R. (1988). DNA-bound Fos proteins activate transcription in yeast. Cell 52 , 179-184.
Ma, J., and Ptashne, M. (1987). A new class of transcriptional activators. Cell 51 , 113-119.
Marcus, S., Polverino, A., Barr, M., and Wigler, M. (1994). Complexes between STE5 and components of the pheromone-responsive mitogen-activated protein kinase module. Proc. Natl. Acad. Sci. USA 91 , 7762-7766.
Mendelsohn, A. R., and Brent, R. (1994). Applications of interaction traps/two-hybrid systems to biotechnology research. Curr. Op. Biotechn. 5 , 482-486.
Printen, J. A., and Sprague, G. F., Jr. (1994). Protein-protein interactions in the yeast pheromone response pathway: Ste5p interacts with all members of the MAP kinase cascade. Genetics 138 , 609-619.
Reymond, A., and Brent, R. (1995). p16 proteins from melanoma-prone families are deficient in binding to Cdk4. Oncogene 11 , 1173-1178.
Rose, M., and Botstein, D. (1983). Construction and use of gene fusions to lacZ (beta-galactosidase) that are expressed in yeast. Methods Enzymol 101 , 167-80.
Thomas, B. J., Gunning, D. A., Cho, J., and Zipursky, L. (1994). Cell cycle progression in the developing Drosophila eye: roughex encodes a novel protein required for the establishment of G1. Cell 77 , 1003-1014.
Tjian, R., and Maniatis, T. (1994). Transcriptional activation: A complex puzzle with few easy pieces. Cell 77 , 5-8.
Van Aelst, L., Barr, M., Marcus, S., Polverino, A., and Wigler, M. (1993). Complex formation between RAS and RAF and other protein kinases. Proc. Natl. Acad. Sci., U.S.A. 90 , 6213-6217.
Yuan, Y. O., Stroke, I. L., and Fields, S. (1993). Coupling of cell identity to signal response in yeast: interaction between the alpha 1 and STE12 proteins. Genes Dev 7 , 1584-97.
Zervos, A. S., Gyuris, J., and Brent, R. (1993). Mxi1, a protein that specifically interacts with Max to bind Myc-Max recognition sites. Cell 72 , 223-232.
Last modified: Thursday, July 24, 2003 2:43:49 PM