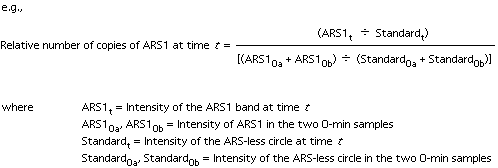Replication timing by density transfer
互联网
Replication timing by density transfer
M. K. Raghuraman
1. Grow cells at least 7 generations in
- 0.1% (w/v) [13 C]- acetate or glucose
- 0.01% (w/v) [15 N]- ammonium sulfate
- Amino acids/supplements (as required, added to the same concentration as in normal synthetic medium)
--dissolved in "-N" medium and filtered. (I add 0.1 mg/ml ampicillin if I'm feeling paranoid.)
The doubling time of RM14-3a is ~ 2.6 hr at 23o C in glucose and 6-8 hr (depending on aeration) in acetate--longer if you're selecting for a plasmid. RM14-3a is MATa, cdc7-1ts , bar1, trp1-289, leu2-3,112, ura3-52, his6 in an A364A background.
I aim for about 32 x 106 cells per time point. That gives enough DNA for at least 3 blots, leaving enough leeway for errors.
2. When the cell density is 2 x 106 cells/ml (OD660 ~ 0.16), arrest with 200 nM alpha factor (1-1.25 doubling times, until % unbudded cells stops increasing). NOTE: This alpha factor concentration is for bar1 strains. BAR1+ strains require 15x more alpha factor.
3. Filter, wash in Ycomp+alpha factor, resuspend in Ycomp+[12 C]-glucose+ alpha factor.
[I don't particularly try to resuspend the cells in exactly the same volume they were in originally, but rather, in a volume that's convenient.]
4. Transfer to 37o C, wait for culture to reach 37o C, then add Pronase (0.01-0.05 mg/ml final conc.). [You can add dry Pronase. I usually dissolve the Pronase in 5 ml of minimal medium, just to avoid having the dry Pronase stick to the sides of the flask.]
5. Hold at 37o C for 90-120 min (until budded cells accumulate). Remove one sample (t = 0), then swirl the flask in ice-water and return it to 23o C. Continue to collect samples (until t = ~80 min).
The ice-water treatment is to quick-chill the culture to 23o C, and is usually for ~1 min. You can empirically determine how long it takes to bring your desired volume of medium from 37o to 23o C.
6. Collecting samples: Freeze 8 ml of 0.1% Na-azide, 0.2 M EDTA in "50 ml" Oakridge-type plastic, screw-capped centrifuge tubes (frozen slanted to maximize the surface area). During the experiment, mix 20 ml of cells with 1/50 volume of 10% Na-azide in a 35 ml Corex tube (on ice) and immediately transfer the mix to a tube of frozen EDTA/azide. Alternatively, squirt the 10% azide on the frozen azide/EDTA and immediately add the cell sample. Vortex or shake the tube vigorously to chill the sample and break up the frozen EDTA. Spin down the cells in a chilled centrifuge, wash with 1 ml cold water per sample in Eppendorf tubes and freeze the pellets at -20o C.
7. Extract DNA ( smash-and-grab with glass beads), digest with the enzyme of choice in 0.1 ml of reaction mix per sample. [I resuspend the nucleic acids pellet in 0.05 ml of 10 mM Tris, 0.1 mM EDTA, and add an equal volume of a 2x enzyme mix that contains the appropriate restriction enzyme buffer, restriction enzyme (usually 1 microliter per sample) and 0.25 microgram/ml RNase A. If the DNA prep is clean, the digest can go overnight at 37 degrees C.]
Choosing a restriction enzyme: the fragment to be probed should ideally be 3-15 kb , to avoid broad, overlapping heavy-heavy and heavy-light DNA peaks in the CsCl gradient.
8. Check for completion of the digest by running 8-10 microliters of it on a gel. To be certain, the gel should be blotted and probed for a known fragment. However, if you are familiar with the pattern expected for a particular restriction enzyme (e.g., EcoRI), you may be able to decide just from the ethidium bromide-stained gel whether the digest was complete. I generally blot and probe the gel anyway.
CsCl gradients
9. Mix each sample (90-92 microliters, restriction enzyme and all) with 9.141 g of CsCl solution, to give a refractive index of 1.4042-1.4043. (Remember, it's a lot easier to dilute a solution that's too dense than to make a too-dilute solution more dense!)
CsCl solution :
Use 1.28 g dry CsCl per gram of 10 mM Tris, 100 mM EDTA , pH 7.5. The T10 E100 should have a refractive index of 1.3395. It may too high when you first make it (up to 1.4010 or thereabouts); just add 10 mM Tris until the refractive index comes down. After the CsCl is in solution, the refractive index should be 1.4052. It's worth adding 90 microliters of dummy restriction enzyme mix to 9.141 g of the CsCl solution just to make sure that you get the right refractive index. This dummy CsCl mix then serves to top off the real samples.
Note: Calibrate to a refractive index of 1.3330 for distilled water. 9.141 g CsCl solution = 5.25 ml. I find that I get much more consistent readings by weighing out the CsCl solution (in small, sterile culture tubes) than by pipetting it out. I initially used 5.4 ml CsCl (= 9.402 g; DNA scaled to match), but that leaves about 0.25-0.3 ml solution left over after the Quickseal tubes are filled. With 5.25 ml CsCl + 0.1 ml DNA, you may find that there is a fair-sized bubble at the top of the Quickseal tube. This is okay--use the dummy mix to bring up the volume and balance the tubes.
10. Spin at 50 000 rpm for 18 hr, 28 000 rpm for 3.5 hr, no brake (i.e., deceleration=0) at either step (use dual program).
11. Fractionate the gradients (10 drops per fraction, collected in multi-well microtitre plates), read the refractive index of every 3rd or 4th fraction as soon as possible thereafter.
Slot blots
12. HH DNA peaks at refractive index of about 1.405, and HL at 1.4040, so you should use fractions from ~1.4060-1.4030. I go for 24 fractions (to take up the 24 slots per lane of the slot blot), so you may have to juggle a bit to get the range you want. Alternatively, you could load all the fractions and avoid this problem, but then the slot blot becomes more complex.
Use 42 microliters of each fraction to be blotted, and microtitre plates for the denaturation/neutralizing:
Mix 42 microliters of DNA (in CsCl) with 28 microliters of 1 N NaOH (=> 0.4 N NaOH final conc.). Seal the plate with an adhesive sheet and incubate at 65o C for 1 hr.
Cool to room temperature and neutralize with an equal volume (70 microliters) of 20x SCP . Pre-wet the membrane in 10x SCP. Run 140 microliters of 10x SCP through the slots and then apply the sample. Run another 140 microliters of 10x SCP through--to get the last bit of the sample out of the microtitre well, I first add this 10x SCP to the well where the sample used to be, then apply it to the slot blot. Remove the membrane and UV crosslink immediately. I let cross-linked membranes sit in 6x SCP, 1% Sarkosyl, 0.1% BSA (diluted from Calbiochem's 30% solution) while the next ones are being done.
Note: I find that I can save on yellow tips by using the same set (on a multichannel pipettor) several times, rinsing the tips out by pipetting up and down with water (in a Pyrex dish) between samples. (I've done the test of rinsing and then using the same tips to apply 10x SCP to a lane of the slot blot; I got no hybridization there, indicating that there was no carry-over of sample. You may choose to use fresh tips--there was one night when I had the last box of tips and was forced to improvise!)
13. Pre-hybridize and hybridize as usual.
Timing markers
We routinely use two or three sequences as our markers for early and late replication. Previously, we used to cut plasmid DNA to obtain fragments to label as probes. These days, we use a set of PCR primers and yeast genomic DNA to amplify the fragments of interest. Our standard marker sequences and their PCR primers are:
ARS305 (chromosome III, very early-replicating, may show some "escape" replication at the cdc7 block; see Reynolds et al., 1989, Mol. Cell. Biol . 9 :4488 ).
- Primers:
- 5'-ATTCGCCTTCTGACAGGACG-3'
- 5'-ATAACGGAGACTGGCGAACC-3'
GAL3 (chromosome IV, ARS1-adjacent, early-replicating; see McCarroll and Fangman, 1988, Cell 54 :505).
- Primers:
- 5'-CCTACATGGCCATAGATCCG-3'
- 5'-AAGGTGGATAGTGCAACCGC-3'
R11 (chromosome V, late-replicating; see Ferguson et al., 1991, Cell 65 :507).
- Primers:
- 5'-GATTGCACGACCAACTTCGG-3'
- 5'-GTTAGGTAGAGCAGGCAGAC-3'
Data analysis
The quantitation method described below is based on Apple Macintosh applications and file formats; comparable options may be available on the Windows platform as well.
14. The hybridization on the slot blot is quantitated by some means (we use a Packard InstantImager or a Molecular Dynamics PhosphorImager), and the hybridization intensities so obtained are plotted for each time point (i.e., each gradient).
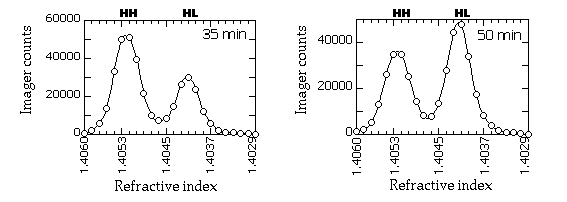
Typical gradient profiles look something like this (in this example, the slot blots were probed with a RAP1 fragment and quantitated on a PhosphorImager):

15. The plot for each time point is saved as a PICT file (if you use KaleidaGraph, a set of plots can be saved as one PICT file).
16. The HH and HL peaks are quantitated by measuring the area under each peak. The PICT files are opened in NIH Image , and the area under each peak is measured by tracing the peak with the freehand tool and applying the "Measure" command. Because the HH and HL peaks tend to overlap, you will have to exercise some judgement in tracing the overlapping portions of the cureves. We use the outer half of each peak as a guide and trace a symmetrical line down the overlapping portion. An example of the curves we would typically trace:

17. The extent of replication at each time point is calculated from the equation
![]()
- Catalog # 28,201-4 Sodium acetate-1,2-13 C2 99% atom purity, $242 for 1 gram
- Catalog # 38,937-4 D-Glucose-13 C6 99% atom purity, $193 for 1 gram
- Catalog # 29,928-6 Ammonium-15 N2 -sulfate 98% atom purity, $83 for 1 gram
- All from Sigma-Aldrich (previously from Isotec, which is now part of Sigma-Aldrich)
"-N" medium =1.61 g yeast nitrogen base w/o amino acids and ammonium sulfate, 11.1 g succinic acid, 6.67 g NaOH per litre.
Pronase : We use Calbiochem's Pronase (catalog #53702), which seems pretty clean--i.e., plates spread after Pronase treatment aren't contaminated--but I heard from someone who'd used Sigma's Pronase and had massive growth of Streptomyces.
20x SCP = 0.6 M Na2 HPO4 , 2.0 M NaCl, pH 6.8
Add to 350 ml dH2 O:
- 18.61 g Na2 EDTA?H2 O
- 1.5 g NaOH pellets
- 5 ml 1M Tris-HCl pH 7.5
Stir until the solids have dissolved, bring the pH to 7.5 with concentrated NaOH (<0.5 ml), and bring the volume up to 500 ml with dH2 O. Filter-sterilize; do not autoclave. The refractive index of the solution should be 1.3393-1.3395.
CsCl : from Gallard-Schlesinger Industries, Carle Place, NY 11514-1731. It's 99.9% pure "Special Biochemical Grade", Catalog # 612501, $150/kilogram.