PCR技术的原理与方法
互联网
PCR定义
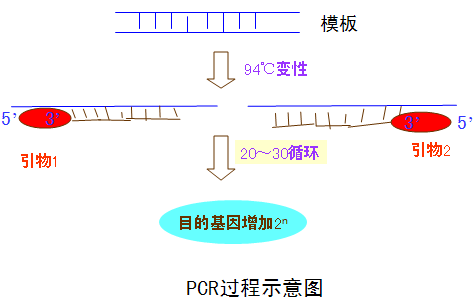
PCR(Polymerase Chain Reaction)即聚合酶链式反应,是指在DNA聚合酶催化下,以母链DNA为模板,以特定引物为延伸起点,通过变性、退火、延伸等步骤,体外复制出与母链模板DNA互补的子链DNA的过程。是一项DNA体外合成放大技术,能快速特异地在体外扩增任何目的DNA.可用于基因分离克隆,序列分析,基因表达调控,基因多态性研究等许多方面。
PCR技术的基本原理
一、PCR反应成分
1、模板DNA;
2、引物;
3、四种脱氧核糖核苷酸;
4、DNA聚合酶;
5、反应缓冲液、Mg2 等。
二、PCR反应基本步骤:
1、变性:高温使双链DNA解离形成单链(94℃,30s)。
2、退火:低温下,引物与模板DNA互补区结合(55℃,30s)。
3、延伸:中温延伸。DNA聚合酶催化以引物为起始点的DNA链延伸反应(70~72℃,30~60s)
以上述三个步骤为一个循环,每一循环的产物均可作为下一个循环的模板,经过n次循环后,目的DNA以2n的形式增加。
PCR反应的成分和作用
总体积:一般为25μl~100 μl
一、无Mg2 buffer:由纯水、kcl、Tris组成。Tris用于调节反应体系pH值,使Taq酶在偏碱性环境中反挥活性。kcl可降低退火温度,但不能超过50 mmol/L,否则会抑制DNA聚合酶活性。
二、Mg2 :终浓度为1.5~2.0mmol/L,其对应dNTP为200 μmol/L,注意Mg2 与dNTPs之间的浓度关系,由于dNTP与Taq酶竟争Mg2 ,当dNTP浓度达到1 mmol/L时会抑制Taq酶的活性。 Mg2 能影响反应的特异性和产率。
三、BSA:一般用乙酰化的BSA,起着减少PCR管对Taq酶的吸附作用,对Taq酶有保护作用。
四、底物(dNTPs):dNTPs具有较强酸性,其储存液用NaOH调pH值至7.0~7.5,一般存储浓度为 10 mmol/L,各成份以等当量配制,反应终浓度为20~200μmol/L.高浓度可加速反应,但同时增加错误掺入和实验成本;低浓度可提高精确性,而反应速度会降低。
五、Taq酶:能耐95℃高温而不失活,其最适pH值为8.3~8.5,最适温度为75~80℃,一般用72℃。能催化以DNA单链为模板,以碱基互补原则为基础,按5’→3’方向逐个将dNTP分子连接到引物的3’端,合成一条与模板DNA互补的新的DNA子链。无3’→5’的外切酶活性,没有校正功能。某种dNTP或Mg2 浓度过高,会增加其错配率。用量一般为0.5~5个单位/100μl.
六、模板:PCR对模板DNA的纯度不要求很高,但应尽量不含有对PCR反应有抑制作用的杂质存在,如蛋白酶、核酸酶、TqaDNA聚合酶抑制剂、能与DNA结合的蛋白质。
模板DNA的量不能太高,否则扩增可能不会成功,在此情况下可适当稀释模板。
七、引物:引物浓度一般为0.1~0.5μmol/L,浓度过高会引起错配和非特异扩增,浓度过低则得不到产物或产量过低。引物长度一般15~30个碱基,引物过长或过短都会降低特异性。其3’末端一定要与模板DNA配对,末位碱基最好选用A、C、G(因T错配也能引发链的延伸)。引物G C约占45~55%,碱基应尽量随机分布,避免嘧啶或嘌呤堆积,两引物之间不应有互补链存在,不能与非目的扩增区有同源性。
PCR反应条件的选择(影响因素)
一、温度参数:
1、变性:模板变性完全与否是PCR成功的关键,一般先于94℃(或95℃)变性3~10min,接着94℃变性30~60s.
2、退火:退火温度一般低于引物本身变性温度5℃。引物长度在15~25bp可通过公Tm=(G C)×4℃ (A T)×2℃计算退火温度,一般退火温度在40~60℃之间,时间为30~45s.如果(G C)低于50%,退火温度应低于55℃。较高的退火温度可提高反应的特异性。
3、延伸:延伸温度应在Taq酶的最适温度范围之内,一般在70~75℃。延伸时间要根据DNA聚合酶的延伸速度和目的扩增片段的长度确定,通常对于1kb以内的片段1min是够用的。
二、循环数:
PCR的循环数主要由模板DNA的量决定,一般20~30次循环数较合适,过多的循环数会增加非特异扩增产物,具体要多少循环数可通过预试验确定。
PCR产物积累规律:
反应初期产物以2n呈指数形式增加,至一定的循环数后,引物、模板、DNA聚合酶形成一种平衡,产物进入一个缓慢增长时期(“停滞效应”),即“平台期”。到达平台期所需PCR循环数与模板量、PCR扩增效率、聚合酶种类、非特异产物竟争有关。
PCR常见问题
一、没有扩增产物
1、循环温度:变性温度、退火温度
2、引物设计
3、DNA聚合酶活性
4、抑制性成份(蛋白酶、核酸酶、其它抑制聚合 酶活性的成份)
5、DNA样品
二、非特异产物及电泳呈涂布状
1、Mg2 浓度
2、调整引物、模板、聚合酶的用量
3、适当减少循环数
4、适当提高退火温度,缩短退火或延伸时间
三、引物二聚体的形成
1、检查引物的序列
2、提高退火温度
3、调整引物与模板浓度
4、增加引物长度
四、假阳性结果的预防
1、实验器材应一次性使用(吸咀、PCR管)
2、注意操作环境,戴一次性手套
3、严格PCR操作规程
4、多次取样的试剂应分装
5、设置阴性对照和阳性对照
PCR相关技术
一、锚定PCR(anchored PCR):
引进锚定引物,可以帮助克服序列未知或序列未全知的障碍。在DNA3’-末端加上poly(dG)尾,与此相对应的锚定引物poly(dC)一般应在十二聚以上。
二、不对称PCR(asymmetric PCR)
不对称PCR主要是在PCR体系中设计不同的引物浓度,两条引物浓度比为1:50或1:100,前12个循环两条模板等量扩增,之后低浓度引物消耗殆尽,其扩增产物减少以至于无;而高浓度引物介导产生的扩增产物(即单链DNA)逐渐增加,可得到大量单链DNA(ssDNA)用于直接测序。
三、反向PCR(inverse PCR)
四、多重PCR(multiplex PCR)
多重PCR是用多对引物同时对模板DNA上的多个区域进行扩增。
多重PCR技术的难点不是在于其原理和操作的复杂性,而是在于其多对引物的设计,必需保证多对引物之间不形成引物二聚体,引物与目标模板区域具有高度特异性。
应用于基因诊断,对与疾病相关的基因(庞大)进行扩增检测。
五、逆转录PCR(reverse transcription PCR,RT-PCR)
由mRNA逆转录产生cDNA链作为PCR反应模板。逆转录合成cDNA时,引物可选用特异引物、随机六聚体引物或寡聚dT(12-18)。设计RT-PCR的引物时最好是分散在不同的外显子上,以免基因组DNA的污染导致假阳性结果
六、差别PCR(differential PCR)
差别PCR是将靶基因与已知拷贝的参照基因置于同一PCR反应体系中,用同一套引物进行扩增,电泳染色后可根据参照基因的扩增产物与待测基因扩增产物的相对丰度对待测基因进行定量。
七、原位PCR(In situ PCR)
原位PCR是指在组织或细胞标本片上直接进行PCR,对细胞中的靶DNA进行扩增,通过掺入标记基团直接显色或结合原位杂交进行检测的方法。可分为直接法和间接法。
基本步骤:组织切片或细胞固定→蛋白酶消化→原位PCR扩增→冲洗→产物检测
优点:灵敏度高,可进行细胞内定位
八、荧光定量PCR(FQ-PCR)
荧光定量PCR:融汇PCR技术、DNA探针杂交技术(标记有荧光报告基团和荧光淬灭基团),结合先进的光谱检测技术发展起来的一项新技术。
主要原理是在待扩增区域结合上DNA探针,PCR过程中,具有5’→3’外切酶活性的Taq酶延伸引物链到DNA探针时,将DNA探针逐个降解,释放出荧光报告基团,这样PCR体系中荧光的强度与PCR产物量之间存在正比关系,可通过测定荧光强度而对PCR产物定量。
荧光定量PCR的优点:
1、可进行准确的定量检测。用于基因诊断。
2.、定量范围宽。
3、特异性更强,克服了假阳性。
4、操作简单快速,无须后处理和电泳检测。
5、安全,技术易于学习,易于进行电脑化数据管理。
PCR技术的应用
1、遗传性疾病的基因诊断
2、传染病的诊断(肝炎病毒)
3、癌基因检测
4、法医学(亲子鉴定)上的应用
5、DNA测序
6、基因克隆
7、引入基因点突变,基因融合等