材料与仪器
SSC cDNA阵列 dNTP 混合液 dCTP 酵母 tRNA DNA 聚合酶 I
Qiaquick PCR 纯化试剂盒 BioPrime 标记试剂盒 Micrcon 30 滤器 杂交炉 Maxiprep DNA 提取试剂盒
步骤
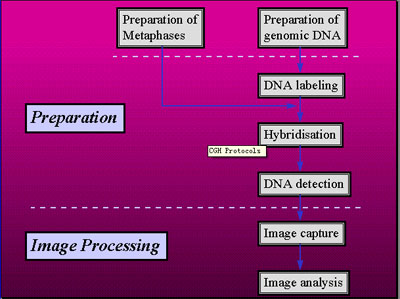
一、cDNA阵列CGH
(1)阵列制备
与变性探针杂交前,先用封闭液(盖上盖片)在37℃封闭cDNA阵列1h
(2)随机引物标记基因组DNA
1.肿瘤和正常组织基因组DNA各2μg分别用Dnp II消化1〜1.5h。将消化产物进行纯化(QiaquickPCR试剂盒)、真空干燥,用25μL水重悬。
2.使用Bioprime标记试剂盒基本按说明书进行随机引物标记,稍作改动。100℃变性DNA和20斗随机引物(试剂盒中含有)5min,迅速放于冰上冷却,并加人2.5μL dNTP、1.25μL dCTP、IμL Cy3/Cy5-dCTP、IμL Klenow片段(试剂盒中含有),37℃孵育90min。
3.混合Cy3和Cy5标记产物,上样到Micrcon30滤器。2000g离心10min,检查样品容器确定标记产物的存在(紫色)。直接加入30μL Cot-1DNA、100μg酵母tRNA、20μg poly(dA-dT),5000g离心20min。为了重新收集样品,加入15ml杂交缓冲液,将Micrcon30滤器倒置放入新的收集管,16000g离心1min。
(3)探针变性和杂交
1.在加热的水浴锅或PCR仪上100℃变性探针90s,冰上冷却探针后,在37℃复性0.5〜1h。
2.将探针加到微阵列上,盖上玻璃盖片并用橡皮泥封片。在65℃用杂交缓冲液湿润的湿盒中杂交16〜20h。
(4)洗脱
1.将cDNA微阵列于2×SSC、0.03%SDS中65℃洗5min,接着用1×SSC、0.2×SSC室温下各洗脱5min。
2.低速(50g)离心5min使标本片干燥。
二、阵列CGH
(1)阵列制备
1.基因组克隆(BAC、PAC、黏粒等)在相应抗生素存在的条件下生长,并用商业化试剂盒大量提取基因组DNA,—般得到10μg数量级的产物。用酚/氯仿的标准化程序进一步纯化DNA。
2.DNA大小和质量在1%琼脂糖凝胶电泳上评估,用UV光度计定量DNA。
3.靶DNA经超声波破碎得到1.5〜15kb片段,沉淀、重悬至适宜浓度,用毛细管以200〜400μm直径点样到玻璃载片上。
4.向阵列上加上20μL封闭液,盖上盖片,37℃湿盒复性1h。
(2)用缺口平移法标记基因组DNA制备探针
1.肿瘤和正常组织基因组DNA各2μg分别用缺口平移法进行标记,反应混合物如下:
A) Cy3反应物(加水至总体积100μL
(1)肿瘤基因组DNA:2μg。
(2)10×Cy3dNTP:10μL。
(3)DNA聚合酶1μL。
(4)DNaseI。
B) Cy5反应物(加水至总体积100uL):
(1)正常人基因组DNA:2μg。
(2)10×Cy5dNTP:10μL。
(3)DNA聚合酶1μL。
(4)DNaseI。
2.16℃标记反应1.5h后,将标记反应混合物放于冰上。
3.标记产物的大小在1%琼脂糖凝胶电泳进行评估,进行CGH实验的理想片段长度在500〜2000bp范围内。如片段过长,在反应混合物中加入适量的DNA聚合酶I和DNaseI,在16℃继续反应。
4.向标记反应混合物中加入0.1倍体积的0.3mol/LEDTA终止反应。
5.用Sephade×G50离心柱除去标记混合物中未掺入的核苷酸。
6.混合两种标记探针,加人50μL Cot-1DNA、0.1倍体积的3mol/L乙酸钠、2倍体积的冰冷100%乙醇沉淀探针。用70%乙醇洗涤沉淀、晾干,然后用20uL杂交缓冲液重悬探针。
(3)探针变性和杂交
1.75℃变性探针5min,然后在37℃复性0.5〜1h以充分封闭重复片段。
2.将探针加到完成预复性的阵列上,盖上盖片并橡皮泥封片,在37℃用杂交缓冲液湿润的湿盒中杂交24h。
(4)洗脱
1.阵列在55℃50%甲酰胺/2×SSC,pH7.0中洗3次,每次10min,然后在含0.1%NP-40,pH8的0.1mol/L磷酸钠缓冲液中室温洗5〜10min。
2.去掉多余的液体,加DAPI/抗淬灭剂后盖上盖玻片。
注意事项
2.在自动化和实用的批量提取方法出现之前,许多实验室采用大量提取试剂盒提取靶DNA制备微阵列CGH。这是一种费时费力的实验,靶DNA在多次的阵列点样后用完时,需要反复多次的提取DNA。如果有足够的纯化靶DNA(用大量或小量制备,至少应有几百个纳克模板DNA),DOP-PCR可用来提供无限的靶DNA。
3.在整个杂交过程中,确保微阵列湿润是至关重要的。随时加入杂交缓冲液以确保杂交盒湿润,以免探针或封闭混合物蒸发。如果微阵列干了,结果将不可信。
4.这里所列的是对在大约(18X16)mm2范围内达到3500个样的cDNA微阵列的优化实验方案。当采用较大点样区域内的较高密度的微阵列时,DNA量和最后的杂交体积必须按比例提高。
5.为得到高质量的微阵列CGH结果,未标记的基因组DNA片段大小和纯度是非常重要的。用于标记的DNA的纯度越低,得到的微阵列结果背景越高、杂交信号越弱。这里叙述的是从新鲜组织中提取基因组DNA进行微阵列CGH的最优实验方案。
6.选择用限制性内切核酸酶进行消化对提高标记效率非常重要。值得注意的是,降低标记前DNA片段平均长度,将提高标记效率,而过度消化得到的探针片段太小而不能用于与cDNA靶杂交。在我们实验室中,使用EcoRI消化人类基因组DNA得到非常满意的稳定结果。
7.在开始建立实验方法时,进行一系列的杂交和洗脱温度梯度实验,以得到最佳杂交强度和特异性。发现37℃杂交和55℃洗脱可非常灵敏地检测高拷贝获得和扩增,而洗脱温度提高到65℃将降低微阵列的信号强度。过低的洗脱温度可能产生非特异结合(过多的黄色信号)。我们建议预实验中用来源不同的样本DNA进行不同的标记,以得到最佳的实验技术特异性。
8.到目前为止,仍没有标准化的阵列制备方法。发表的论文中靶DNA浓度为400〜10μg/mL时,用人工点样在多聚赖氨酸处理的玻片上,或Zyg/yL靶DNA点样在氨丙基三甲氧硅烷处理过的玻片上[12]。请注意随着机器点样仪的自动化程序的出现,DNA浓度和玻片处理方法都可能随之改变。
9.这里提到的方法是假定在(22X20)mm2玻璃盖片范围内构建最大量的小格化点样面积。另外,假定靶DNA在阵列制备的过程中已经变性。否则,在与探针杂交前,微阵列必须在70%甲酰胺/4×SSC中变性2min
10.探针的最终长度依赖于DNA酶I的浓度。用于CGH实验的探针最佳长度在500〜2000bp范围内。酶的储存液浓度为1×10-4U/μL,实验前用DNA酶I稀释液新鲜配制成终浓度5×10-5U/μL的使用液。然而,随着要求所获得的DNA理想片段长度的改变,酶浓度也相应的做出调整。
11.将0.05〜0.1倍体积的每种标记混合物进行DNA染色(如溴化乙锭)的凝胶电泳。由于可以有助于阵列CGH的问题解决,所以推荐采用琼脂糖凝胶电泳评估标记
来源:丁香实验